
Human Population Genetics and Genomics ISSN 2770-5005
Human Population Genetics and Genomics 2022;2(1):0002 | https://doi.org/10.47248/hpgg2202010002
Review Open Access
Genetics and population history. The case of the Iberian Peninsula and the “origin” of Basques
Jaume Bertranpetit


Academic Editor(s): Guido Barbujani, Lounès Chikhi
Received: Oct 19, 2021 | Accepted: Nov 30, 2021 | Published: Feb 9, 2022
This article belongs to the Special Issue Luca Cavalli-Sforza’s legacy, 100 years after his birth
© 2022 by the author(s). This is an Open Access article distributed under the terms of the Creative Commons License Attribution 4.0 International (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly credited.
Cite this article: Bertranpetit J. Genetics and population history. The case of the Iberian Peninsula and the “origin” of Basques. Hum Popul Genet Genom 2022; 2(1):0002. https://doi.org/10.47248/hpgg2202010002
One of the main challenges of human population genetics has been the reconstruction of the population history of humans at different scales, from the origin of the modern humans to the history of specific groups. In all cases information from other historical sciences (including archaeology, linguistics and physical anthropology) should match in the unique frame of population history. Cavalli-Sforza, had a pioneering role in defining the problem and putting together a database of classical genetic markers and statistical methods to make the genetic approach of high relevance. One of the problems studied refers to the Basque population, establishing its distinctiveness and “origin”. As in many other settings, research in the area in the last few decades has flourished by adding much DNA information and statistical analysis to corroborate or correct the initial hypotheses. In the case of the Basques, the differentiation without strong external genetic influences has been confirmed as due to isolation, and instead of being pre-Neolithic, it is currently dated to the Iron Age, only some 2,500 year ago.
Based on: “Bertranpetit J, Cavalli-Sforza LL. A genetic reconstruction of the history of the population of the Iberian Peninsula. Ann Hum Genet 1991; 55:51-67.”
Keywordspopulation history, basques, Iberian Peninsula, classical genetic markers, genetic structure
The publication of the volume “The History and Geography of Human Genes” by Cavalli-Sforza, Menozzi and Piazza in 1994 [1] was a landmark for a whole new discipline: the application of population genetics to the reconstruction of the evolutionary history of human populations. The scope was global, a reconstruction of the past from the origins of anatomically modern human to the peopling of the continents, and even of specific regions. The analysis reached microscale when the historical questions were relevant enough and, of course, if genetic data were available. The main novelty of Cavalli-Sforza, Menozzi and Piazza's approach was that evidence from different disciplines, historical sciences, physical anthropology, and linguistics was used both to generate hypotheses testable at the genetic level, and to interpret the results of those tests. The basic chain of events that matters in this reconstruction of the past is the demographic history of human populations, a history of people, retrieved through their genes, and thus of births, reproduction and deaths. In turn, these individual processes may lead to population growth or decrease, migration and admixture with recipient populations, patterns of mating, be it through social practices or not, and patterns of reproduction, for which both the average number of children and the differences among couples matter. In fact, by studying patterns of allele frequencies one obtains valuable insights into the demographic history of human populations. Traditionally, the analysis focused on modern human populations, from which we can retrieve the genetic (or nowadays genomic) information; however, in the last years the expansion of ancient DNA techniques has made available a huge amount of genotypes and sequences of archaeological samples, that may go back from a few years ago to fossils hundreds of thousands of years old.
In a sense, starting from the book by Cavalli-Sforza and colleagues, our duty during these years has been to develop new genetics and statistical analyses in order to enhance, confirm, and correct Cavalli-Sforza’s work.
The main questions that genetics can help address have to do with the prehistorical and historical events causing the main social shifts in certain populations. No doubt cultural changes are at the base of the main economic changes, which in turn have a strong influence upon demography. However, archaeological data have little power to discriminate between changes occurring only at the cultural level (e.g., transmission of a new technology from one population to another) and changes entailing demographic processes (e.g., diffusion of a new technology through migration of the people who possessed that technology).
The case of the Neolithic transition is the clearest documented case of the demographic impact of a cultural or economic shift in prehistory. Moving from food collection to food production forever changed how humans live, eat, and interact, paving the way for modern civilization ([2]; see also Barbujani’s contribution in this issue). Now we have even been able to recognize the immunological shift that the cultural change had on our genome in responding to a new set of infectious diseases from the proximity to domestic animals [3]. One of the main methodological problems is that most genetic evidence of population differentiation is extremely difficult to fit into a temporal framework. Only under very strict theoretical models can we, for example, calculate an age for a differentiation process due to isolation between two populations (i.e., by drift). Theory shows that drift is strongly dependent on population size; thus, the smaller the population, the faster the process of population divergence [4]. This is the reason why ancient events, when population sizes of human populations were small, were likely to have left a profound footprint in the genetic differentiation of human populations.
As was written in a rather poetical way, after stating that “the few demographic pieces of information taken from historical sources and compared with linguistic records support the hypothesis that the genetic structure of Italy still reflects the ethnic stratification of pre-Roman times” Piazza et al. (1988) [5] concluded their paper with: “Just as we see the light of the stars coming to us thousands of years after its time of origin, in the same way the genetic structure of contemporary Italy reflects the history of our distant past as if it had lain frozen for centuries”.
Thus as a parti pris in the human population genetic analyses it has been assumed that the oldest demographic events are those that are more likely to have profoundly altered the human genetic landscape and thus the differences we find today have been interpreted as very old events in population history. But it is impossible to infer an exact date from genetic information about modern people; the ancient DNA studies have been essential on giving an evidence of the time frame. We will come back to this point while discussing the time depth of the Basque differentiation.
The linguistic differentiation in humans is a fantastic process in which similarities among languages have been used to recognize similarities interpreted as coming from a common language spoken in the past and having differentiated. This analysis has been able to recognize linguistic families (like Indo-European) or linguistics isolates (like Basque). To put a temporal framework has been (and still is) a matter of much debate, although recently the application of phylogenetic methods has created a quite accurate framework in the case of the Indo-European language family [6]. The main point to consider here is which social structure and which social changes may be at the basis of the rate of language change and language replacement.
It is of special interest to understand the spread of a specific language family as a diversification pattern after an expansion (and usually, extinction) of a previously spoken language. Why some languages last over centuries and others do not is also an interesting question.
The study of human differentiation in morphology, including the classification of people into what were called ‘races’, has been controversial in the reconstruction of human evolutionary history. The main problem with approaches based on morphological traits has been the difficulty of applying phylogenetic analyses to them [7]. In fact, morphology has mostly been used for taxonomic purposes without an underlying evolutionary base. The main problem is the lack of an evolutionary clock for morphological changes, with some traits evolving faster and others slower because of differences in selective pressures. When a phylogenetic approach has been forced onto morphological analyses, the results varied depending on the traits used and the statistical tools. Variations in morphological traits undoubtedly contain important information, but cannot be used as drivers of hypotheses. Moreover morphological variation is of interest on a global or very wide scale, but not within continents.
Similar to the way Cavalli-Sforza analyzed populations at a worldwide scale, many scientists have focused on small-scale analyses, be it for a geographic area or for ethnically interesting groups. In 1991 Luca Cavalli-Sforza and I published a paper on the reconstruction of the population history of the Iberian Peninsula [8] that was later extended to the Pyrenees [9] and southwest Europe [10]. This can be considered the first paper using genetic data for many loci to unravel the population history of the Basques. Others authors had published papers using one or a few genetic markers. There is no doubt that Mourant’s early work [11] on the ABO blood groups paved the ground, by proposing that “the Basques are a relict population which at least in Spain has suffered no significant admixture of elements akin to the general western European population”. Later, with members of his team, the same author recognized the strong differentiation of Basques for the Rh alleles [12], adding support to his proposal.
Other authors had realized that the genetic differences in the known blood groups of Basques compared to neighboring populations suggested some specificity in the population history. In fact, in Bertranpetit and Cavalli-Sforza’s (1991) paper [8], we turned the question upside down by asking: “Is the singularity of Basques going to be confirmed if we take a wider geographic area and use as many genetic markers as were available?” The work consisted of collecting allele frequencies for 20 genetic systems with 54 alleles (34 of which independent) in a total of 635 population studies of the Iberian Peninsula (Spain and Portugal). For each allele, we used a smoothing technique to calculate frequencies for the nodes of a grid and principal component analysis was applied to the complete data matrix. The main point of this work was that Basque differentiation clearly popped up strongly among neighboring populations and (no surprise) blood groups ABO and Rh had a strong correlation with the first principal component, which accounted for 27.1% of the total variation. Thus, the Basque region held a very strong signal of differentiation in the Iberian Peninsula and in a wider European framework (Figure 1).

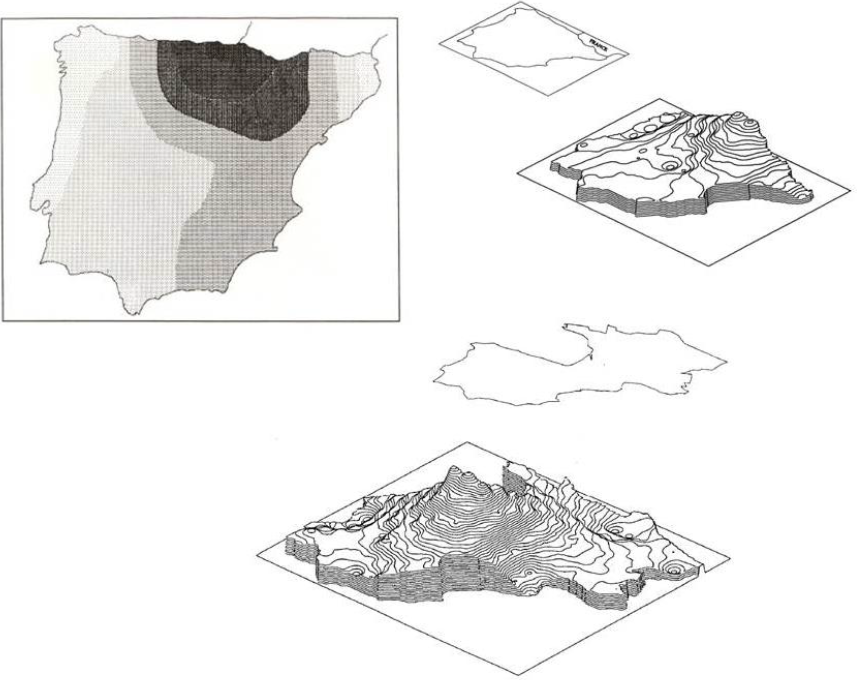
The interpretation of the signal was straightforward, based on prehistory and linguistics: the Basque differentiation was the remnant of a pre-Neolithic differentiation. As we have discussed, the genetic data do not come with a credible time frame, and hence that interpretation had to be based on other fields. We concluded that within the European context, in which the model of a Neolithic wave of advance was gaining support, it seemed adequate to postulate a pre-Neolithic hypothesis of the origin of the Basque population. Accordingly, it was logical to assume that the Basques’ ancestors were already somewhat differentiated in pre-Neolithic times and that the Neolithic wave had less impact than in neighboring populations. In fact, in the Basque region there has been a delay in absorbing most of the cultural innovations, which could have led to a cultural absorption without the population replacement of most of Europe. There has been a durable continuation of special cultural aspects, mainly the language, which are of unquestionable antiquity.
From today’s vantage point, it is interesting to look back to what we learned from the classical genetic studies of population history.
Genetic studies with classical genetic markers have been long questioned because functionally relevant markers may be subject to the action of natural selection; as a consequence, their geographic distribution may not reflect population history, but rather adaptation. The classical response to this objection was that selection would have been acting in different ways for different gene variants and thus, its effect could be compensated by the analysis of many markers. Nonetheless, it is really impossible to rule out that selection has played a role in determining spatial patterns of genetic variation. In addition, the number of markers could not increase beyond the discovery of protein variants, and thus was somewhat limited. Other kinds of genetic markers, in much higher numbers and with clear neutrality had to be uncovered, and the technique of high-throughput DNA sequencing would provide the solution.
For classical genetic markers, different researchers typed different polymorphisms in different populations. A more general way of typing the same set of markers in the same populations was needed.
The use of principal component analysis (PCA) was the primary tool used to extract the maximum amount of information from the genetic variations observed in many different alleles. This method applied to sets of allele frequencies for a set of populations, was fundamental in the initial interpretation of the geography of genetic data. Nonetheless, the method has been criticized [13], mainly because the geographical structure in the data does not necessarily mean that there has been migration. With the advent of SNP (single-nucleotide polymorphism) technologies the use of PCA has flourished more than before, as it certainly is efficient in summarizing genomic information, using it for individuals instead of populations, and projecting them on a 2-dimensional plot. To have a direct plot of individuals with the spread of variation has been the key to the recent success of in-depth genetic analyses.
To solve the problems of using classical genetic markers for population history inference, a new window was opened with the possibility of analyzing regions of the human genome that do not recombine; the main advantage of this technique is that all existing variations were produced by mutations on the same DNA molecule. This approach became possible as human population genetics was embracing a molecular view through a direct interrogation of the DNA sequence by the tedious manual Sanger sequencing technique. Two elements of the human genome were known not to recombine: the mitochondrial genome (mtDNA), which is maternally transmitted and the paternally transmitted NRY (non-recombinant fraction of the human Y chromosome, 95% of its length). In both cases, the initial technical capabilities did not allow full analysis of these regions. Only subsets could be tackled: 360 to 1,000 bp of the mtDNA control region (full analysis of the 16,569-bp mtDNA became routine in the early 2010s) and a few to >50 pre-ascertained SNPs plus 10-20 STRs for the NRY, extended to ~9 Mb of sequence by the late 2010s.
The analysis of mtDNA variation has many advantages, both experimentally (large number of molecules, short length, regions with varying amount of diversity, higher mutation rate than the nuclear genome, etc.) and at the level of bioinformatics analysis, especially for the possibility of applying a gene genealogy approach. In that method, starting from a set of sequence variants, one reconstructs the whole evolutionary tree, including the reconstruction of ancestral, extinct, sequences. Most importantly, trees offer an estimate of the moment in which mutations occurred (with large confidence intervals) even if this becomes computationally very complex, when there is a large amount of data. Comprehensive trees are now available, showing all but the shallowest branches in mtDNA (http://www.phylotree.org, [14]) and Y-chromosome ([15]; tree in Figure 1) genealogies.
But it also has some problems. The mtDNA and NRY are just two genetic loci, which are strongly affected by drift, and maybe by selection, and are a small and biased sample from which to derive general conclusions at a population history level. The age of the nodes of the gene genealogy were taken as references for age of the populations, which often happened long before any demographically relevant event, such as a population expansion. Mutation ages and the ages of such expansions are rarely the same [16]. Thus, while interest in population genetics of non-recombining genome regions is undeniable, their application was marred by many instances of over-interpretation. An additional issue lies in the reification of the deeper branches of the genealogies, the so-called haplogroups, which were often depicted as if they were autonomous evolutionary entities supplanting the actual populations [17]. The discussion of the presence or frequency of particular haplogroups in the Basques lies beyond the scope of this article and would not enrich the discussion.
Studies on uniparental marker diversity in the Basques are abundant, and they have described Basques as wholly European. Sub-branches other than the core European variation are absent in Basques, while other sub-branches are found at high frequencies, some with clear signs of having differentiated in situ. Nonetheless the conclusions of several studies have been driven by the enduring hypothesis of the persistence of a pre-Neolithic background in the people from Basque country. Only a seminal and a more modern study will be discussed here.
The initial study on mtDNA [18] pointed to a remarkable homogeneity of the Basques, with minor differences relative to other European populations, suggesting a strong founder effect, and concluded that “although classical markers, such as blood groups and protein polymorphisms, clearly separate the Basques (and the Sardinians) from other European populations, this distinctiveness was not found using mtDNA”. The ‘haplogroup’ approach opened the field to other population interpretations, many of which turned out to be inaccurate (see Barbujani in this issue). For example, Cardoso et al, (2013) [19] interpreting the haplogroups as “Paleolithic” or “Neolithic” conclude that “these findings provide robust evidence of a partial genetic continuity between contemporary autochthonous populations from the Franco-Cantabrian region, specifically the Basques, and Paleolithic/Mesolithic hunter-gatherer groups…further supporting the notion of a predominant Paleolithic genetic substrate in extant European populations”. Several other papers have proposed similar conclusions although the data were compatible with other interpretations.
The first study on the Y-chromosomes of Basques from a population history view [20] revealed a low Y-chromosome diversity in Basques within the European diversity landscape, which the authors attribute to a low effective population size maintained through generations. They recognized that some Y-chromosome lineages in modern Basques originated in pre-Neolithic times and have been evolving since then. These results were compatible with the general view of a partially ancient background with later migrations and strong drift. Recent studies on Y-chromosomes ([21]and references therein) “support the hypothesis that during the Bronze Age a dispersal of individuals occurred that led to the replacement of the Paleolithic/Neolithic Y-chromosome composition in Western Europe by Indo-European R-S116 lineages.” The interesting point here is that the time of replacement was more recent than previously proposed and this is the main conclusion of the ancient DNA study [22] discussed below. Thus two independent studies converge in indicating that the genetic features of Basques took shape more recently than commonly assumed.
The development of molecular biology technologies allowed a more direct examination of DNA sequences, and the list of genetic markers all along the genome increased. Some studies analyzed the Basque population using Alu insertions and STRs (still of wide use in forensics). They are mainly of historical interest.
SNPs were, and still are, a very powerful tool as genetic markers. A seminal paper [23] analyzed 144 SNPs in Basque DNA samples from France and Spain, as well as from northern and southern Spain and North Africa. They found no genetic differences between Basques and non-Basques, which was a surprising result--but the number of SNPs was low. When the availability of SNP arrays made the large-scale interrogation of SNPs possible, a new era of research was opened in the field. Hundreds of thousands of biallelic markers could be screened in a single experiment, most of them clearly neutral, allowing giant steps forward in the genetic description of populations. In an initial study of Basques [24], instead of typing individuals, pools of DNA of 30 individuals for each of the ten populations studied were considered. Results were clear and the “analysis showed that, when a genome-wide perspective is applied, Basques are not particularly differentiated from other Iberian populations”.
Soon, another paper in the same journal [25] found contradictory results: “we present the first high-throughput analysis including Basques from Spanish and French provinces, and show that all Basques constitute a homogeneous group that can be clearly differentiated from other European populations”. The divergence between the two studies, as seen with many more studies done since then, were likely due to the pooling method of the samples used in the first study and the Basque genetic differentiation has been fully confirmed in whole genome approaches.
In a wide SNP analysis of 1413 individuals from across Spain, [26], described historical events as the main drivers of genetic differentiation, with a clear genetic impact of population movements associated with both the Muslim conquest and the subsequent Reconquista. The distant past and the Basque differentiation was not specifically discussed. But even with these ample sets of markers, analyses were not able to produce a detailed view of the past origins of the population. It was necessary to resort to ancient DNA studies, to be able to describe genetic diversity in multiple prehistoric and historic time periods. We are not going to report the studies of ancient uniparental markers, as they have not provided a clear enough picture of the past population history.
The capability of extracting and sequencing ancient DNA has been a major step forward. It took long time to fully develop these methodologies, mainly due to technical difficulties associated with the DNA extraction, PCR amplification, postmortem DNA damage and contamination [27]. With the advent of new sequencing techniques, methods for the study of DNA in ancient samples have become more standardized and usable by a wider set of scientists [28]. Two main approaches have been competing, based respectively on obtaining whole genome sequences from the old remains, or on typing a set of pre-defined SNPs through what came to be called hybridization capture [29, 30].
The two methods have pros and cons. From a purely scientific point of view, whole genome sequencing is the best option, as in principle this will provide all the available information. However, the enormous cost and difficulties in obtaining them due to contamination and degradation has made other, more limited approaches, technically and economically preferable. Despite recognizing the possible ascertainment bias in the SNPs chosen and the impossibility of analyzing rare variants, hybridization capture is the technique that has allowed, in a short period of time, the acquisition of genetic data from thousands of ancient DNA samples with more to come from old remains.
In the study of Basques, ancient DNA entered the scene with analyses of mtDNA ([31]and several later papers) followed by whole genome sequencing of Neolithic Atapuerca [32] with inconclusive results suggesting that Basques and their language may have been linked to the spread of agriculture during the Neolithic. The main results came from a large study of the Iberian Peninsula mainly done in the lab of David Reich at Harvard [22], with data from 400 individuals covering around 8000 years, from Mesolithic till recent historical times. This study clearly showed that the Basque genetic substratum was not as ancient as previously postulated, but it did confirm the Basques’ genetic isolation since the Bronze Age, ~ 2200-900 BCE. As seen in Figure 2, all populations in the Iberian Peninsula shared a similar population history until then, with first, the Paleolithic ancestry related to the central European hunter-gatherers; second, a very strong influence of the Neolithic wave of advance, as postulated by Cavalli-Sforza; and third, a central European steppe-related ancestry, also referred as the Yamnaya culture, in the Bronze Age. This last population movement into Iberia likely correlated with the introduction of the Urnfield tradition and the Indo-European languages to the region. In Iberia several populations did not adopt an Indo-European language, including the later Iberian cultural groups and what we now call Basque groups; only the latter preserved that language with the spread of Latin and Romance languages at the turn of the era. After this mix, the resulting population had an estimated 40% of steppe-related ancestry and 60% of what already existed. This is the common genetic substrate of all the peoples of the Iberian Peninsula, including the Basques. Later, Basques remained isolated from the gene flow processes that had a strong influence on the rest of the Iberian Peninsula, with influences of the Roman expansion and the Muslim conquest (Figure 2).
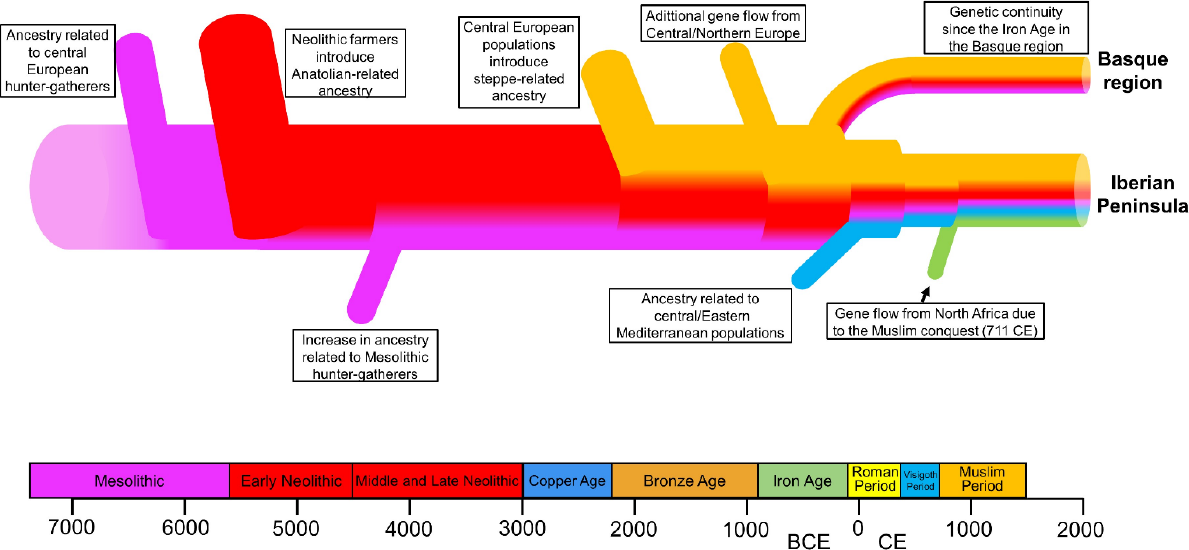
Figure 2 Schematic representation of the migratory events in the Iberian Peninsula over the last 8,000 years, specifically delineating the Basque region. Modified from an original provided to the author by Dr Olalde, following [22].
These findings and interpretations have been confirmed at the whole genome level for the extant population, including a microgeographical analysis of the Basque country (Flores-Bello et al., 2020). In the largest study carried out to date, with DNA samples from almost 2,000 current and ancient individuals and with DNA of 190 people whose four grandparents were born in the same area, the genetic uniqueness of the Basques in Europe has been confirmed. More to the point, this uniqueness is not due to an extraordinary origin, but simply to greater isolation since the Iron Age, some 2,500 years ago.
This study clearly confirmed that Basques were not simply a continuation of the hunter-gatherers of pre-Neolithic times nor of Neolithic times, but of the most recent Iron Age populations. The analysis performed at a microgeographical level showed a correlation between the historical geographic distribution of Euskara (the Basque language) and the genetic heterogeneity detected nowadays in the area. This suggests that language could have acted as a cultural barrier that facilitated the isolation of the Basques since the Iron Age, for example during Roman or Muslim rule. The preservation of these genetic characteristics can be seen as the modeled influence of Iron Age ancestry in southwestern Europe, where it had a strong correlation with the Basque area (Figure 3). Thus the genetic specificity of the Basques can be viewed as a peak in what is the present Basque country with clines into the southwest and the northeast (shown in different colors in Figure 4).
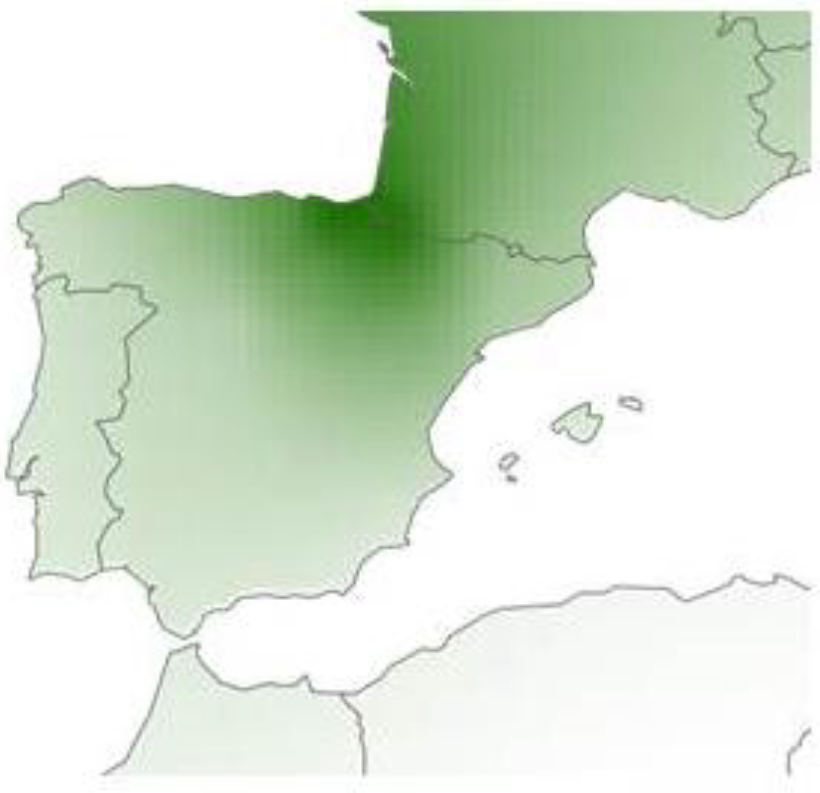
Figure 3 Modeled influence of the Iron Age-related ancestry in SW Europe, where it had a strong correlation with the Basque area. Original provided to the author by Dr. Flores-Bello [33].
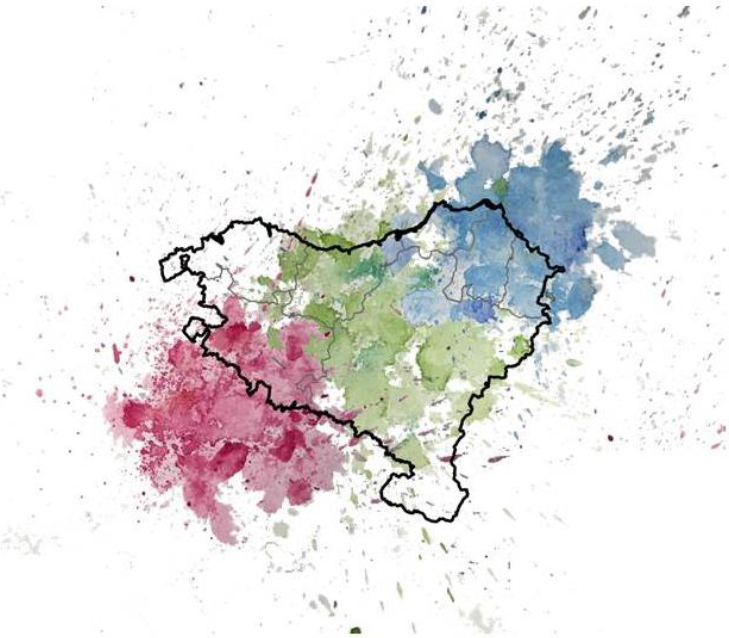
Figure 4 Artistic representation of the genetic structure in the historically Basque-speaking territory, with green symbolizing Basque ancestry and blue and red, the mixture with the surrounding populations. Drawing by André Flores-Bello, kindly offered for publication.
Luca Cavalli-Sforza has had a unique influence on the field of human population genetics. Many of his ideas that nowadays are considered mainstream, were still in their infancy or did not exist thirty years ago. They include very general topics, such as the basics of how genetics can recover human (demographic) history or the recognition and measure of the factors underlying genetic differences among populations. It is fair to give credit mainly to him for the multidisciplinary approach to the study of human evolution, using data and concepts from genetics, demography, anthropology, archaeology, and linguistics. Cavalli-Sforza developed his multidisciplinary view thanks to a deep knowledge of all aspects of population genetics, including many new developments both in molecular and biostatistical techniques. One key aspect was the importance of using phylogenetic trees to understand genetic variation, as can be seen in Figure 5, a picture of a paper tablecloth that we had in the cafeteria during a working lunch and in which he depicted the main points of our conversation in 1990.

Figure 5 Paper tablecloth with the discussion points drawn and written by Cavalli-Sforza in 1990. Kept and pictured by the author.
When the book he would call “librone” was published [1], many researchers (including myself) realized that a new area was coming, expanding the genetic markers used, increasing sampling populations and employing sophisticated methods to test his proposals, correct them and extend them based on new findings. This has been the case for the genetic singularity of the Basques and its interpretation in terms of population history in relation to cultural events. During the thirty years after the seminal papers on the Basque singularity and our collaborative paper on the study of classical genetic markers, many relevant works have been published. The main picture of Basque genetic singularity has not changed (in spite of a few papers that claimed it did not exist), with drift and isolation still considered the main producers of their differentiation. What has changed lately is the exact placement in time of the demographic processes leading to the Basques’ isolation not in pre-Neolithic times, but 2,500 years ago in the Iron Age.
Jaume Bertranpetit is a member of the Editorial Board of the journal Human Population Genetics and Genomics. He was not involved in the review or decisions related to this manuscript.
This study has been possible thanks to grant PID2019-110933GB-I00/AEI/10.13039/501100011033 awarded by the Agencia Estatal de Investigación (AEI), Ministerio de Ciencia, Innovación y Universidades (MCIU, Spain) and with the support of Secretaria d’Universitats i Recerca del Departament d’Economia i Coneixement de la Generalitat de Catalunya (GRC 2017 SGR 702). Part of the “Unidad de Excelencia María de Maeztu”, funded by the AEI (CEX2018-000792-M).
Thanks to several colleagues for discussion and help: Francesc Calafell (uniparental markers), David Comas (whole genomes of Basques) André Flores-Bello (Figures 3 and 4), Iñigo Olalde (ancient DNA and Figure 2) and Guido Barbujani.
| 1. | Cavalli-Sforza LL, Menozzi P, Piazza A. The History and Geography of Human Genes. Princeton, New Jersey: Princeton University Press; 1994. |
| 2. | Bellwood P. First farmers. The origins of agricultural societies. Malden, USA: Blackwell; 2005. |
| 3. | Domínguez-Andrés J, Kuijpers Y, Bakker OB, Jaeger M, Xu CJ, van der Meer JWM, et al. Evolution of cytokine production capacity in ancient and modern European populations. eLife. 2021;10:e64971. [Google Scholar] |
| 4. | Wright S. Classification of the factors of evolution. Cold Spring Harbor Symp Quant Biol. 1955;20:16-24. [Google Scholar] |
| 5. | Piazza A, Cappello N, Olivetti E, Rendine S. A genetic history of Italy. Ann Hum Genet. 1988;52:203-213. [Google Scholar] [CrossRef] |
| 6. | Gray R, Atkinson Q, Greenhill S. Language evolution and human history: what a difference a date makes. Phil Transac R Soc B. 2011;366:1090-1100. [Google Scholar] [CrossRef] |
| 7. | Bertranpetit J. The genome over time and evolution as a strategy for the endurance of life. Memòries de la Reial Acadèmia de Ciències i Arts de Barcelona (RACAB). 2021;1063:47. (Article in Catalan). [Google Scholar] |
| 8. | Bertranpetit J, Cavalli-Sforza LL. A genetic reconstruction of the history of the population of the Iberian Peninsula. Ann Hum Genet. 1991;55:51-67. [Google Scholar] |
| 9. | Calafell F, Bertranpetit J. Mountains and genes: population history of the Pyrenees. Hum Biol. 1994a;66:823-842. [Google Scholar] |
| 10. | Calafell F, Bertranpetit J. Principal component analysis of gene frequencies and the origin of Basques. Am J Phys Anthrop. 1994b;93:201-215. [Google Scholar] [CrossRef] |
| 11. | Mourant AE. The blood groups of the Basques. Nature. 1947;160:505. [Google Scholar] |
| 12. | Chalmers JNM, Ikin EW, Mourant AE. The ABO, MN and Rh blood groups of the Basque people. Am J Phys Anthropol. 1949;7:529-544. [Google Scholar] [CrossRef] |
| 13. | Novembre J, Stephens M. Interpreting principal component analyses of spatial population genetic variation. Nat Genet. 2008;40:646-649. [Google Scholar] [CrossRef] |
| 14. | van Oven M, Kayser M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum Mutat. 2009;30:E386-E394. [Google Scholar] [CrossRef] |
| 15. | Jobling MA, Tyler-Smith C. Human Y-chromosome variation in the genome-sequencing era. Nat Rev Genet. 2017;18:485-497. [Google Scholar] [CrossRef] |
| 16. | Degnan JH, Rosenberg NA. Discordance of species trees with their most likely gene trees. PLoS Genet. 2006;2:762-768. [Google Scholar] [CrossRef] |
| 17. | Barbujani G, Goldstein DB. Africans and Asians abroad: genetic diversity in Europe. Annu Rev Genom Human Genet. 2004;5:119-150. [Google Scholar] [CrossRef] |
| 18. | Bertranpetit J, Sala J, Calafell F, Underhill PA, Moral P, Comas D. Human mitochondrial DNA variation and the origin of Basques. Ann Hum Genet. 1995;59:63-81. [Google Scholar] [CrossRef] |
| 19. | Cardoso S, Villanueva-Millán MJ, Valverde L, Odriozola A, Aznar JM, Piñeiro-Hermida S, et al. Mitochondrial DNA control region variation in an autochthonous Basque population sample from the Basque Country. Forensic Sci Int Genet. 2012;6(4):e106-e108. [Google Scholar] [CrossRef] |
| 20. | Alonso S, Flores C, Cabrera V, Alonso A, Martín P, Albarrán C, et al. The place of the Basques in the European Y-chromosome diversity landscape. Eur J Hum Genet. 2005;13:1293-1302. [Google Scholar] [CrossRef] |
| 21. | Luis JR, Palencia-Madrid L, Mendoza VC, Garcia-Bertrand R, de Pancorbo MM, Herrera RJ. The Y chromosome of autochthonous Basque populations and the Bronze Age replacement. Sci Rep. 2021;11(1):5607. [Google Scholar] [CrossRef] |
| 22. | Olalde I, Mallick S, Patterson N, Rohland N, Villalba-Mouco V, Silva M, et al. The genomic history of the Iberian Peninsula over the past 8000 years. Science. 2019;363:1230-1234. [Google Scholar] |
| 23. | Garagnani P, Laayouni H, González-Neira A, Sikora M, Luiselli D, Bertranpetit J, Calafell F. Isolated populations as treasure troves in genetic epidemiology: the case of the Basques. Eur J Hum Genet. 2009;17:1490-1494. [Google Scholar] [CrossRef] |
| 24. | Laayouni H, Calafell F, Bertranpetit J. A genome-wide survey does not show the genetic distinctiveness of Basques. Hum Genet. 2010;127:455-458. [Google Scholar] [CrossRef] |
| 25. | Rodríguez-Ezpeleta N, Alvarez-Busto J, Imaz L, Regueiro M, Azcárate MN, Bilbao R, et al. High-density SNP genotyping detects homogeneity of Spanish and French Basques, and confirms their genomic distinctiveness from other European populations. Hum Genet. 2010;128:113-117. [Google Scholar] [CrossRef] |
| 26. | Bycroft C, Fernandez-Rozadilla C, Ruiz-Ponte C, Quintela I, Carracedo A, Donnelly P, Myer S. Patterns of genetic differentiation and the footprints of historical migrations in the Iberian Peninsula. Nat Commun. 2019;10:551. [Google Scholar] [CrossRef] |
| 27. | Krause J, Pääbo S. Genetic time travel. Genetics. 2016;203:9-12. [Google Scholar] |
| 28. | Stoneking M, Krause J. Learning about human population history from ancient and modern genomes. Nat Rev Genet. 2011;12:603-614. [Google Scholar] [CrossRef] |
| 29. | Burbano HA, Hodges E, Green RE, Briggs AW, Krause J, Matthias Meyer M, et al. Targeted investigation of the Neandertal genome by array-based sequence capture. Science. 2010;328:723-725. [Google Scholar] [CrossRef] |
| 30. | Fu Q, Meyer M, Gao X, Stenzel U, Burbano HA, Kelso J, Pääbo S. DNA analysis of an early modern human from Tianyuan Cave, China. Proc Natl Acad Sci U S A. 2013;110:2223-2227. [Google Scholar] [CrossRef] |
| 31. | Izagirre N, de la Rúa C. An mtDNA analysis in ancient Basque populations: implications for haplogroup V as a marker for a major paleolithic expansion from southwestern Europe. Am J Hum Genet. 1999;65:199-207. [Google Scholar] [CrossRef] |
| 32. | Günther T, Valdiosera C, Malmström H, Ureña I, Rodriguez-Varela R, Sverrisdóttir OO, et al. Ancient genomes link early farmers from Atapuerca in Spain to modern-day Basques. Proc Natl Acad Sci U S A. 2015;112:11917-11922. [Google Scholar] [CrossRef] |
| 33. | Flores-Bello A, Bauduer F, Salaberria J, Oyharçabal B, Calafell F, Bertranpetit J, et al. Genetic origins, singularity, and heterogeneity of Basques. Curr Biol. 2021;31:2167-2177. [Google Scholar] [CrossRef] |

![]()
Copyright © 2026 Pivot Science Publications Corp. - unless otherwise stated | Terms and Conditions | Privacy Policy




