
Human Population Genetics and Genomics ISSN 2770-5005
Human Population Genetics and Genomics 2023;3(2):0004 | https://doi.org/10.47248/hpgg2303020004
Original Research Open Access
Mutation Rate (Under)estimation through Mendelian Incompatibilities
Antonio Amorim
1,2,3


Academic Editor(s): Joshua Akey
Received: Mar 1, 2023 | Accepted: Jun 1, 2023 | Published: Jun 10, 2023
© 2023 by the author(s). This is an Open Access article distributed under the Creative Commons License Attribution 4.0 International (CC BY 4.0) license, which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is correctly credited.
Cite this article: Amorim A. Mutation Rate (Under)estimation through Mendelian Incompatibilities. Hum Popul Genet Genom 2023;3(2):0004. https://doi.org/10.47248/hpgg2303020004
Mutation is the key player in evolutionary change, both in micro and macro scenarios. Defined as any hereditary change in the transmitted genome from progenitor to offspring, it embraces a heterogeneous set of phenomena, in terms of molecular bases and consequences. Here we limit our analyses to those corresponding to site-specific changes in nucleic acid sequence composition, without alteration in the genome size, i.e., substitutions. These analyses will be performed in the framework of the classical Mendelian diploid mode of transmission, without sex-linkage, under a simple Hardy-Weinberg model. Under these conditions, we show that: (i) per site mutation rates are evolutionarily uninformative, biallelically defined parameters being required to predict the genetic behavior of the population and are also essential to medical and forensic applications; (ii) at polymorphic sites (heterozygosity > 0), estimates of mutation rates through the simple counting of Mendelian incompatibilities in pedigrees are always underestimations, depending heavily on the allelic frequencies, a fact that may lead to erroneous evolutionary inferences, such as that (iii) apparently equal forward and reverse mutation rates, as estimated from Mendelian incompatibilities, do not equate to stable equilibrium. An approach allowing to correct the underestimation, by adopting a sequential strategy of allele specific estimation is presented.
Keywordsmutation rate estimation, substitution, population genetics, evolution
Any hereditary change in the transmitted genome (DNA or RNA) from progenitor to offspring, is called a mutation. In multicellular, sexually reproducing organisms, if the copying error happens in the formation of gametes the resulting offspring will carry a germinal mutation, and from now on, mutation will be used in this sense. For simplicity’s sake, in the analyses below only substitutions, single site modifications not involving deletion or insertion will be used, and the diploid homogametic mode of transmission is assumed, under the Hardy-Weinberg model, except for the occurrence of mutation. The simplest system (two alleles per site) is used, multiallelic situations being tractable in a sequential fashion, using the biallelic approach, by choosing one allele at a time and pooling all the others.
The occurrence of mutations (and the estimation of their frequency or mutation rate) can be inferred from phylogenetic (e.g., [1,2]), or from genealogical studies. There are two main types of genealogical studies, according to the kind of family material used: progenitor/offspring duos, or progenitors (father and mother)/offspring trios. With this sort of family material, which avoids the complexity of multigeneration analyses [3], copy errors can be identified (and quantified) through the observation of Mendelian incompatibilities, i.e., instances in which (i) an allele present in the offspring is absent from both parents, or, (ii) when the offspring does not show an allele necessarily transmitted by the progenitor.
Many genetic and environmental factors are known to influence mutation rate, such as progenitors’ age, or sequence context, but those effects will be ignored in the analyses below, which aim specifically to evaluate and correct the biases involved in the estimation of mutation rates via the Mendelian incompatibilities approach.
Detection of mutations via the Mendelian incompatibilities approach has been already criticized, as it ignores the possible occurrence of hidden events [4–9]. We will briefly explain the rationale behind this claim for the two types of family material (duos or trios) mostly used in this detection method. It is must be made clear that mutations can only occur hiddenly at polymorphic sites (heterozygosity > 0), which constitute a small fraction of the genome. The situation for duos is summarized in Table 1. There, the codominant alleles are coded as 1 and 2, with respective frequencies p, and q, as above, with m denoting the per site mutation rate (i.e., of allele 1 into 2 and the reciprocal, assumed to be equally frequent). It is common, however, to disregard the possible heterogeneity of ‘forward’ and ‘reverse’ mutation frequencies and to calculate the overall mutation rate per site, symbolized as m, so firstly we will carry the analysis under this framework. Expected frequencies at each cell are computed assuming that HW conditions are met, except for mutation. Note that using duos, separate analyses for male and female progenitors can be made, allowing the estimation of sex-specific mutation rates, or pooled, assuming they are equal.
Table 1 Expected genotype frequency distribution of parent/offspring duos with mutation. Mutation rate is m; allele frequencies are p and q, for alleles 1 and 2, respectively; shaded cells correspond to the cases of detectable Mendelian incompatibilities.

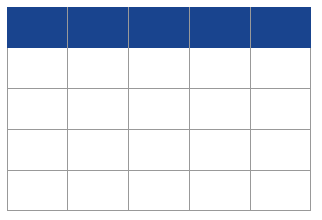
The estimation of the mutation rate under this approach is simply the frequency of the observed incompatibilities. Denoting their frequency, relative to the total number of studied pairs, as O1-1/2-2, and O2-2/1-1 (for the Mendelian incompatible pairs 1-1/2-2, and 2-2/1-1, respectively, corresponding to the shaded cells in Table 1), the estimate of rate of the mutation per site is
The inspection of the table shows however that many more mutations are possibly occurring, although not causing any Mendelian incompatibility. This is the case, for instance, of the duo 1-1/1-2 in which the known progenitor may have transmitted a mutated allele (1 into 2) and the gamete from the unknown progenitor carries allele 1. As these hidden mutations are not detected, we must resort to expected values; the derivation of the required formulas is straightforward and allows us not only to confirm that the above method delivers an underestimate of the mutation rate [10], but also the magnitude of the underestimation. In the simple case above, the expected proportion of observed incompatibilities (corresponding to O1-1/2-2+O2-2/1-1) is (p2qm + q2pm)= pqm. Hence, the corrected rate of the mutation,
Not surprisingly, the value of the deviation from the naïve estimate varies with the allele frequencies, as shown in the Table 2, reporting the results of a simulation, arbitrarily fixing mutation rate value and calculating the expected frequency of Mendelian incompatibilities under various allele frequencies scenarios (calculated according to Equation 2).
Table 2 Variation of the expected frequencies of Mendelian incompatibilities in various allele frequencies scenarios, under a constant per site mutation rate, m = 0.01.
Note that the ratios between the ‘true’ value and the estimate are the same, whatever the value attributed to the mutation rate and take the value of the product of allele frequencies. The conclusion is that the degree of underestimation depends on the allele frequencies, so that the higher the asymmetry between alleles (i.e., the lower the heterozygosity), the greater the underestimation of mutation rate.
This parameter (average mutation rate per site) is, however, of little evolutionary relevance ([11], as well as in forensics: see [12–14]), as it does not allow us to determine if the population is in equilibrium, or if allele frequencies will change, possibly reaching fixation. The relevant parameters enabling this prediction are the allele specific mutation rates, m1>2 and m2>1, and the analysis of the same data from this point of view is shown in Table 3.
Table 3 Expected genotype frequency distribution of parent/offspring duos under allele specific mutations. Mutation rates are m1>2 and m2>1 for transitions of allele 1 into 2 and 2 into 1, respectively; allele frequencies are p and q, for alleles 1 and 2, respectively; shaded cells correspond to the cases of Mendelian incompatibilities.
Then, it would follow the estimates of
and the expected proportion of observed incompatibilities for transitions of allele 1 into 2 (corresponding to O1-1/2-2) would be p2qm1>2, and for the symmetrical 2 into 1 (corresponding to O2-2/1-1), pq2m2>1. That would lead to the conclusion that the Mendelian incompatibilities estimates, not only grossly underestimate the real value but also provide erroneous evolutionary forecasts, as evidenced in Table 4 (computations according to Equations 3 and 4).
Table 4 Variation of the expected frequencies of Mendelian incompatibilities (MI) with the allele frequencies, under equal mutation rates, m1>2= m2>1= 0.01.
This conclusion is however erroneous, as it was based on a fallacious reasoning. In fact, since the mutation rates are now conditioned on the original allele, it is mandatory to analyze the data separately for each allelic case. This analysis will be exemplified with allele 1 in Table 5.
Table 5 Expected genotype frequency distribution of parent/offspring duos involving original allele 1. Mutation rate is m1>2 for transitions of allele 1 into 2; allele frequencies are p and q, for alleles 1 and 2, respectively; shaded cells correspond to the cases of Mendelian incompatibilities.
Now, the shaded cell corresponds to the only case of observable Mendelian incompatibility 1-1/2-2, and the estimation of the mutation rate must consider only the total n° of duos in which allele 1 is transmitted (mutated or not). This total amounts to those with progenitor’s genotype 1-1 (in which allele 1 is the only possible source), with frequency p2, and to those with genotype 1-2, (in which allele 1 as source amounts to ½ of the cases), with frequency pq (adding up to p2+pq = p(p+q)= p). Thus, to the observed frequency of incompatible duos (O1-1/2-2), corresponds the expected value of p2qm1>2/p, and the corrected mutation rate estimate is
Not unexpectedly, making again the comparison between the ‘true’ value and the mutation rate estimated through Mendelian incompatibilities, underestimation is also observed, at the same ratio as for the average per site, mutation rate, as seen in Table 6.
Table 6 Variation of the expected frequencies of Mendelian incompatibilities with allele 1 frequency, under a constant mutation rate = 0.01 of allele 1 into 2.
In all cases, the degree of underestimation depends on the allele frequency of the allele under consideration, so that, the higher the frequency of the selected parental allele, the greater the underestimation of mutation rate.
Performing now the same analysis using trios, and assuming that paternal and maternal mutation rates are equal, the results are summarized in the Table 7.
Table 7 Distribution of mutations occurring in father/mother/offspring trios. Transmission with mutation is denoted by m and without by n; shaded cells correspond to the cases of Mendelian incompatibilities.
It is obvious (although not generally acknowledged, when performing Mendelian incompatibilities estimations), that in this approach each offspring results from two known meiosis [15] and thus the total n° of observations is the double of the sampled trios. This is the only way to correctly accommodate the instances of mutation occurring in both parents, as in trios 1-1/1-1/2-2, or, expectedly, in some of 1-1/1-2/2-2 trios, for instance, in which the Mendelian incompatibility can be attributed to either a single 1n2 paternal mutation or to two 122 (paternal and maternal) mutations.
Consequently, it is impossible to classify these observations as corresponding to one or two mutations, and thus to apply a simple counting method for the estimation of the mutation rates, without assuming a (pseudo) maximum parsimony approach, disregarding the possibility of double mutations in trios like 1-1/1-2/2-2. Therefore, it is impossible to evaluate in the same simple manner, as it was for duos, the proportion of hidden mutations. The issue has been addressed by Slooten and Ricciardi [16] and in their study on the proportion of hidden mutations, the formula derivation (shown in the paper’s Appendix A) assumes no double mutation.
Furthermore, since we now know both parents’ genotypes, the possibility of the other mutation, from allele 2 into 1, must be taken in consideration, which means that while desiring to estimate m1>2, another unknown parameter besides the one we would like to estimate, namely m2>1 must obligatorily to be estimated, since we do not know the original alleles.
Therefore, despite containing much more information than available in duos, the identification of paternal and maternal origin of mutations is much more difficult, and the number of possible ways (with and without mutations) resulting in the same specific genotypic configuration of the trios increases. Even assuming that paternal and maternal mutation rates are equal, the method of counting Mendelian incompatibilities faces serious difficulties.
Since it has already been shown that the estimation of mutations frequency requires allele defined parameters, i.e., not the global, per locus rate of changes, but specifically of each allele into another, we would examine the same type of data under this perspective, searching for the estimation of 112 mutation rate and thus to the subset of trios in which allele 1 is present in either mother, father or both. The corresponding trimming provides however almost no simplification, since only three cells at the lower right corner of Table 6 are removed (those corresponding to the trios in which both parents do not possess allele 1).
Slooten and Ricciardi [16] found that the correlation between the probability of heterozygosity per locus and the frequency of hidden mutations for duos is almost perfect, while in trios was substantially smaller. Therefore, we will not dwell into the full analysis of the trios’ material, safely assuming that, although weaker than in duos, the underestimation of the frequency of mutation is also present at trios and follows similar patterns to those observed in duos.
Mutations are essential for evolutionary change, but the comprehension of their consequences and the development of adequate models and predictions has been hampered by imprecision of the theoretical formulation as well as by the deficient methodology of estimation of their frequency.
We have illustrated the systematic underestimation of the mutation rates when equating the occurrence of mutation with their visibility, by equating mutations to Mendelian incompatibilities. Next, we will show that the usual per site approach is misleading, and the parameter mutation rate requires the definition of the initial allelic state, as well as the resulting one.
Reexamining the results shown in Table 5, we notice that the degree of underestimation of the frequency of mutation involving a specific allele depends on the frequency of the allele itself, so that the higher the allele frequency, the greater the proportion of hidden mutations. This means that unless two alleles are equifrequent, their apparent mutations rates estimated as being equal, may truly be widely different, a difference which can exceed an order of magnitude, depending on the asymmetry between the respective allele frequencies. Inversely, their mutation rates being apparently different may reflect just their different allele frequencies. This is numerically explored in the following table, using the calculations already performed for duos, explained above. There, we have shown that the Mendelian incompatibilities approach would lead to the estimates m1>2=O1-1/2-2/p and m2>1=O2-2/1-1/q, the corresponding expected value being pqm1>2 in both cases. In case m1>2= m2>1, the (under)estimates would obviously take the same value, whatever the allele frequencies. What happens when they are different is numerically illustrated in Table 8.
Table 8 Variation of the expected frequencies of Mendelian incompatibilities (MI) with allele frequencies. Ratio= MI estimate/”true” mutation rate.
If allele specific mutation rates estimation is corrected for the frequency of the parental allele, there is no qualitative difference between the evolutionary predictions made using any of them, since the ratio between the MI estimates and the true value are constant. Nevertheless, due to the general underestimation intrinsic to MI method, the inferred rate of evolutionary change is slower than that of the real one. Note that this conclusion does not contradict the study by Antão-Sousa et al. [14], who ascertained that the estimations of per site mutation rates depend on allele frequency distribution. Indeed, if an allele specific mutation rate estimation is not corrected for the frequency of the parental allele, the Mendelian incompatibilities method provides (under)estimates that may differ by one order of magnitude.
Another misconception consists in assuming that if forward and reverse mutation rates are equal (m1>2=m2>1), then current allele frequency is unchanged over generations. In fact, equilibrium is only reached when genes are equifrequent. It suffices to recall that the frequency, for instance, of allele 1, being p, in one generation would take the value p(1-m1>2)+(1-p)m1>2 in the next.
The major problem with the correct estimation of allelic specific mutation rates is obviously the rarity of cases (both in trios and duos) in which the parental allele can be identified, which implies that these estimates of a rare parametric value will be subject to serious sampling fluctuations. This difficulty does not justify in any case the persistence of the use of per site mutation rates, which may be, for any site with heterozygosity > 0, useless, or even misleading in evolutionary studies as well as in forensic or medical applications.
The differences in phylogenetic versus pedigree-based estimates of mutation rates are among the most puzzling questions in population genetics (e.g., [9,17,18]). Our analyses do not allow to conclude that the differences between the two types of estimates are entirely due to the underestimation of genealogical studies described here. In fact, there are two sources of difficulties arising when trying to compare the values from the two sources. Firstly, most, if not all, phylogenetic estimates involve positions which are (currently) monomorphic in the species analyzed, but with distinct fixed alleles. Of course, in the case of (intraspecifically) monomorphic sites, all mutations are detected in family studies. On the other hand, most of recent literature reporting experimental results on the topic is based on NGS and consequently has a great concern with genotyping quality, and therefore data are analyzed simultaneously in terms of genotyping errors and (true) Mendelian inconsistencies. It turns out therefore difficult to disentangle in these publications the results specifically concerning the latter factor. For example: Bergeron et al. [19] only sites in which both parents were homozygous for the reference allele, and the offspring was heterozygous, were counted; Douglas et al. [20], under a random-allele–error model, reported detection rates of 51%–77% for multiallelic markers and 13%–75% for biallelic markers; and Gordon et al. [21] estimated a detection rate range between 25 and 30%, the detection rate being lowest when the two alleles have equal frequencies; and true genotyping error rate roughly 3.3–4 times that of the apparent error rate at an SNP locus. In a previous work [14], we have obtained similar results: using real population data and simulating mutations in 1,000,000 parent–child duos and parents–child trios, the proportion of hidden mutation was found to vary from 0.143 to 0.546 in duos and from 0.102 to 0.269 in trios, less polymorphic markers showing the higher values.
Although not providing an argument based on new experimental data, I deem to have expanded the research on the topic, providing some new cross-references from different fields of research which, despite dealing with the same problem hardly communicate. I hope to have provided the outline of an approach that may mitigate the difficulties, by adopting a sequential strategy of allelic specific mutation rate estimation which, at the same time, by increasing the statistical power, enhances the sampling efficiency of a rare phenomenon.
Not applicable.
Not applicable.
Not applicable.
This work was partially financed by FEDER: Fundo Europeu de Desenvolvimento Regional funds through the COMPETE 2020: Operacional Program for Competitiveness and Internationalization (POCI), Portugal 2020, and by Portuguese funds through FCT: Fundação para a Ciência e a Tecnologia/Ministério da Ciência, Tecnologia e Inovação in the framework of the projects “Institute for Research and Innovation in Health Sciences” (POCI-01–0145-FEDER-007274).
Antonio Amorim is a member of the Editorial Board of the journal Human Population Genetics and Genomics. The author was not involved in the journal’s review of or decisions related to this manuscript. The author has declared that no other competing interests exist.
| 1. | Nachman MW, Crowell SL. Estimate of the mutation rate per nucleotide in humans. Genetics. 2000;156(1):297-304. [Google Scholar] [CrossRef] |
| 2. | Tian X, Cai R, Browning SR. Estimating the genome-wide mutation rate from thousands of unrelated individuals. Am J Hum Genet. 2022;109(12):2178-2184. [Google Scholar] [CrossRef] |
| 3. | Jónsson H, Sulem P, Arnadottir GA, Pálsson G, Eggertsson HP, Kristmundsdottir S, et al. Multiple transmissions of de novo mutations in families. Nat Genet. 2018;50(12):1674-1680. [Google Scholar] [CrossRef] |
| 4. | Chakraborty R, Stivers DN, Zhong Y. Estimation of mutation rates from parentage exclusion data: applications to STR and VNTR loci. Mutat Res. 1996;354(1):41-48. [Google Scholar] [CrossRef] |
| 5. | Dawid AP, Mortera J, Pascali VL. Non-fatherhood or mutation? A probabilistic approach to parental exclusion in paternity testing. Forensic Sci Int. 2001;124(1):55-61. [Google Scholar] [CrossRef] |
| 6. | Brenner CH. Multiple mutations, covert mutations and false exclusions in paternity casework. Int Congr Ser. 2004;1261:112-114. [Google Scholar] [CrossRef] |
| 7. | Vicard P, Dawid AP. A statistical treatment of biases affecting the estimation of mutation rates. Mutat Res. 2004;547(1-2):19-33. [Google Scholar] [CrossRef] |
| 8. | Vicard P, Dawid AP, Mortera J, Lauritzen SL. Estimating mutation rates from paternity casework. Forensic Sci Int Genet. 2008;2(1):9-18. [Google Scholar] [CrossRef] |
| 9. | Scally A. Mutation rates and the evolution of germline structure. Philos Trans R Soc Lond B Biol Sci. 2016;371(1699):20150137. [Google Scholar] [CrossRef] |
| 10. | Fu YX, Huai H. Estimating mutation rate: How to count mutations? Genetics. 2003;164(2):797-805. [Google Scholar] [CrossRef] |
| 11. | Cano AV, Gitschlag BL, Rozhoňová H, Stoltzfus A, McCandlish DM, Payne JL. Mutation bias and the predictability of evolution. Philos Trans R Soc Lond B Biol Sci. 2023;378(1877):20220055. [Google Scholar] [CrossRef] |
| 12. | Pinto N, Gusmão L, Amorim A. Mutation and mutation rates at Y chromosome specific Short Tandem Repeat Polymorphisms (STRs): A reappraisal. Forensic Sci Int. Genet. 2014;9:20-24. [Google Scholar] [CrossRef] |
| 13. | Amorim A, Pinto N. Estimates of mutation rates from incompatibilities are misleading - guidelines for publication and retrieval of mutation data urgently needed. Forensic Sci Int Genet Suppl Ser. 2019;7(1):612-613. [Google Scholar] [CrossRef] |
| 14. | Antão-Sousa S, Conde-Sousa E, Gusmão L, Amorim A, Pinto N. Estimations of mutation rates depend on population allele frequency distribution: The case of autosomal microsatellites. Genes. 2022;13(7):1248. [Google Scholar] [CrossRef] |
| 15. | Jacquard A. The Genetic Structure of Populations. Berlin: Springer; 1974. |
| 16. | Slooten K, Ricciardi F. Estimation of mutation probabilities for autosomal STR markers. Forensic Sci Int Genet. 2013;7(3):337-344. [Google Scholar] [CrossRef] |
| 17. | Scally A, Durbin R. Revising the human mutation rate: Implications for understanding human evolution. Nat Rev Genet. 2012;13(10):745-753. [Google Scholar] [CrossRef] |
| 18. | Ségurel L, Wyman MJ, Przeworski M. Determinants of mutation rate variation in the human germline. Annu Rev Genomics Hum Genet. 2014;15:47-70. [Google Scholar] [CrossRef] |
| 19. | Bergeron LA, Besenbacher S, Zheng J, Li P, Bertelsen MF, Quintard B, et al. Evolution of the germline mutation rate across vertebrates. Nature. 2023;615:285-291. [Google Scholar] [CrossRef] |
| 20. | Douglas JA, Skol AD, Boehnke M. Probability of detection of genotyping errors and mutations as inheritance inconsistencies in nuclear-family data. Am J Hum Genet. 2002;70(2):487-495. [Google Scholar] [CrossRef] |
| 21. | Gordon D, Heath SC, Ott J. True pedigree errors more frequent than apparent errors for single nucleotide polymorphisms. Hum Hered. 1999;49(2):65-70. [Google Scholar] [CrossRef] |

![]()
Copyright © 2026 Pivot Science Publications Corp. - unless otherwise stated | Terms and Conditions | Privacy Policy




