
Human Population Genetics and Genomics ISSN 2770-5005
Human Population Genetics and Genomics 2025;5(1):0002 | https://doi.org/10.47248/hpgg2505010002
Original Research Open Access
Human adaptation in the Andes Mountains
Jessica De Loma
1
 ,
Mário Vicente
2,3,4
,
Noemi Tirado
5
,
Franz Ascui
6
,
Luis A. Parada
7
,
Jacques Gardon
8
,
Carina Schlebusch
2,9,10
,
Karin Broberg
1
,
Mário Vicente
2,3,4
,
Noemi Tirado
5
,
Franz Ascui
6
,
Luis A. Parada
7
,
Jacques Gardon
8
,
Carina Schlebusch
2,9,10
,
Karin Broberg
1
Correspondence: Karin Broberg
Academic Editor(s): Maanasa Raghavan
Received: Oct 9, 2024 | Accepted: Feb 13, 2025 | Published: Feb 28, 2025
© 2025 by the author(s). This is an Open Access article distributed under the terms of the Creative Commons License Attribution 4.0 International (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly credited.
Cite this article: De Loma J, Vicente M, Tirado N, Ascui F, Parada LA, Gardon J, Schlebusch C, Broberg K. Human adaptation in the Andes Mountains. Hum Popul Genet Genom 2025; 5(1):0002. https://doi.org/10.47248/hpgg2505010002
Humans have adapted to live in diverse environments worldwide. For example, humans living in the Andes Mountains of South America face challenging conditions including high altitude, arid climate, and high concentrations of toxic elements such as arsenic in the soil and water. However, genomic studies of natural selection in indigenous populations in South America are rare and focused mainly on adaptation to high altitude. Here, we conducted a genome-wide search for additional traits showing evidence of positive selection in three ethnic groups from the Andes Mountains, one from the Argentinean Puna (Atacameño-Kolla) and two from the Bolivian Altiplano (Uru and Aymara-Quechua), whose settlements share similar geological characteristics. We identified signals of positive selection in each population by three independent selection scan methods and compared these signals across populations. The three populations showed overlapping selection signals for 116 genes, among these, the most prominent categories, although not significant after multiple testing, were genes involved in sperm motility, opioid signaling and morphine addiction, and pathways related to immune defense against pathogens and cardiovascular functions. Taken together, the results of our multiple genome-wide selection scan methods identify potential positively selected traits that are shared across these three native Andean populations, thus revealing common mechanisms of adaptation in different ethnic groups living in the Andes.
Keywordsselection, native American, opioid, pathogen, high altitude, South America
The Andes Mountains, at an average altitude of 4,000 meters, are the longest mountain range in the world and one of the harshest environments inhabited by humans. The peopling of the Andean highlands is suggested to have occurred around 12,000 years ago, shortly after humans arrived in South America [1]. While migrating to and across the Andes, humans encountered extreme habitats characterized by high altitude, new pathogens, and a very arid climate. Today, the genomes of Andean indigenous people narrate their past evolutionary history, making the Andes a unique setting in which to study human adaptation.
Previous studies of human populations that settled in the Andes have mainly focused on adaptation to high altitude [2–8]. However, the Andean highlands are also characterized by a unique geochemistry that results in elevated concentrations of toxic elements in drinking water, food, and dust. Human adaptation to arsenic, one of the most toxic elements in the Earth’s crust, was recently described in the Argentinean Andes (Puna region) and the Bolivian Andes (Altiplano region), where elevated arsenic concentrations are often found in drinking water [9,10]. However, little is known about other potential selective pressures in the Andes and the genetic history of people populating the region [11].
In this study, we searched for genomic signatures of selection in three native populations of the Andes: the major populations in the Bolivian Altiplano, i.e., the Uru and Aymara-Quechua ethnicities, and the Argentinean indigenous population from San Antonio de los Cobres (SAC) (likely of Atacameño-Kolla descent). Using multiple genome-wide selection scan methods, we aimed to identify potential positively selected traits that are shared across these three native Andean study populations.
The Andean study groups have been described in detail [10,12]. The individuals from the Argentinean Andes were recruited for a study on genetics for arsenic metabolism and sampled from San Antonio de los Cobres (SAC; 3,800 m above sea level in the Puna region) in 2008. Before the recruitment, an information meeting with the health personnel at the local hospital was held. During interviews, the participants were asked, as an inclusion criterion, whether they were originally from SAC and whether their families had lived in the same region for at least 2–3 generations. Directly asking about ethnicity was avoided since it was a potentially sensitive topic in this region at the time of recruitment. Based on later research in the same area, however, people from SAC identified themselves as of Atacameño-Kolla origin [13]. The Atacameños have lived in this region for 11,000 years [14], and traces of human settlements in this area date back to 1,500 years BCE [15]. Since the participants did not identify themselves as belonging to a specific ethnicity at the time of the recruitment, we refer to this population based on their geographic location.
In the Bolivian Andes, we recruited individuals for a study on genetics for arsenic metabolism from ten villages around Lake Poopó, located in the southern part of the Bolivian Altiplano (3,686 m above sea level). Recruitment took place between 2015 and 2018 and involved a series of pre-information meetings with the health personnel and the local authorities, followed by information meetings with the health personnel at the time of recruitment. The study participants were identified as belonging to the Aymara-Quechua or Uru ethnicity based on the location of residency. The Aymara and Quechua are the predominant indigenous groups in this region. They are fairly similar from a genetic standpoint [16,17], and especially around Lake Poopó, the two ethnicities cohabit. Therefore, both ethnicities were assessed as a common study group. In contrast, the Uru communities around Lake Poopó, specifically of Uru-Murato descent, have been historically isolated from other communities in that region, and the number of Uru individuals living in this region is low [18]. We recruited participants from all Uru communities living around Lake Poopó, but not from all Aymara-Quechua communities.
The study areas in Bolivia and Argentina were revisited in 2020 to obtain oral and written informed consent from the participants for this study on whole-genome selection. In total, 81 individuals from the Bolivian Altiplano (65 Aymara-Quechua women, 13 Uru women, and 3 Uru men) and 32 women from the Argentinean Puna agreed to participate in the current study. Most study participants were women, as the men were often away from home to work. All samples and genetic data had been previously obtained and described [9,10]. This study was approved by the Comisión Provincial de Ética del Ministerio de Salud de Salta (Argentina), the Comité Nacional de Bioética (Bolivia), and the Regional Ethic Committee of Karolinska Institutet (Sweden, No. 2020-00495 and 2020-00493).
To return results to the indigenous communities who have contributed to the present study, a series of workshops will be held in several communities. In addition to describing the purpose and methodologies used, the workshops will also be devoted to contextualizing how this genetic research can be understood within their Andean culture.
Study participants were genotyped as described previously [9,10]. Briefly, DNA was extracted from whole blood samples or buccal cells with EZNA Blood DNA Mini kit (Omega Bio-teck, USA) or Qiagen Blood Mini kit (Qiagen, Germany). Genome-wide genotyping was performed at the SNP&Seq Technology Platform in Uppsala (Sweden) on the Illumina Infinium Omni5Exome and on the Illumina Infinium Omni5M bead chips for the Bolivian and Argentinean study groups, respectively. The data was aligned to the human reference genome build, version 37 (hg19).
Using PLINK v.1.90 [19], we filtered for a 5% genotype missingness threshold for SNPs, a 15% genotype missingness threshold for individuals, indels, and duplicates. Further, A/T and C/G SNPs were removed to avoid strand issues when merging to comparative datasets. This quality filtering was performed independently on the Bolivian and Argentinean datasets. Only autosomal SNPs were included.
Principal component analysis and ADMIXTURE were previously described [9] and included in this work in Figure S1 as a reference. Briefly, this dataset included the three Andean populations (Aymara-Quechua, Uru, and SAC) merged with other South American populations reported by Barbieri et al. (2019) [20] (n = 175) and complemented with selected populations of Native American ancestry from North, Central, and South America (n = 312) from a previously published dataset assembled by Lazaridis et al. (2014) [21]. A Hardy-Weinberg equilibrium filter (p-value < 0.001) was used to account for genotyping errors in the Bolivian and Argentinean datasets. First-degree relatives, as determined with the software KING v.2.1.4 [22], were excluded. In total, this dataset had 236,127 autosomal SNPs.
For principal component and admixture analyses [9], we further pruned SNPs in high linkage disequilibrium (plink --indep-pairwise 200 25 0.4), resulting in 120,384 autosomal SNPs. Principal component analysis was done using the smartpca program included in EIGENSOFT [23,24] with default settings. Admixture fractions were inferred using ADMIXTURE [25], with 2–15 clusters (K = 2–15) and 50 replicates, and visual inspection was performed with PONG [26].
Runs of homozygosity were detected in the study participants from the current work. We measured the lengths of homozygosity with PLINK using the following parameters: --homozyg --homozyg-window-kb 5000 --homozyg-window-het 1 --homozyg-window-threshold 0.05 --homozyg-kb 500 --homozyg-snp 25 --homozyg-density 50 --homozyg- gap 100. We summed the total length of homozygous blocks (binned by fragment sizes) for each individual, and then averaged the results by population.
The Andean study populations were merged with selected populations from the 1000 Genomes project (Han Chinese, CHB; Colombians, CLM; Mexicans, MXL; Peruvians, PEL; and Puerto Ricans, PUR) as assembled previously [9]. To assess possible genotyping errors, a Hardy-Weinberg equilibrium filter (p-value < 0.001) was used for each subpopulation. To avoid removing potential selection candidates, which are expected to be out of equilibrium, we excluded only those SNPs that were out of Hardy-Weinberg equilibrium across the four populations from South America, those out of equilibrium common to Aymara-Quechua and Uru, and those out of equilibrium common to Aymara-Quechua and SAC. In total, 1,451,199 autosomal SNPs were kept.
Haplotypes were phased using fastPHASE v.1.4.0 with one random start for 25 iterations and 25 clusters. To aid the phasing, relatives were included in this step. Once haplotypes were phased, to avoid introducing bias due to over-representation of family genotypes during selection scans, first- and second-degree relatives identified with KING were excluded.
To compute the ancestral state of each allele, hg19-aligned versions of the reference genomes of chimpanzee, gorilla, and orangutan were used as previously described [9,10]. For each SNP, an allele was defined as ancestral if it was present in three of the outgroup genomes. The dataset was then filtered based on this information, with only SNPs that were common in the three outgroups retained. SNPs that differed between the outgroups were excluded from the dataset because their ancestral state was uncertain [9].
The integrated haplotype score (iHS) [27] and cross-population extended haplotype homozygosity (XP-EHH) [28] are statistics used to detect positive selection in the genome. These tests are based on the fact that extended haplotype homozygosity arises when allele frequencies increase faster than the rate at which recombination can break them down [29]. XP-EHH detects alleles that are near fixation in one population compared to another population. We compared our three Andean study groups to PEL. The iHS and XP-EHH computations were done using the R package rehh [30], with a maximum distance between two SNPs of 200,000 bp to avoid spurious signals in genomic regions with low SNP density.
The locus-specific branch length (LSBL) test is based on fixation index (FST) values that identifies SNPs with allele frequency differences between three populations [31]. Using Weir and Cockerham’s equation, FST distances measure the differentiation between populations based on the heterozygosity of each SNP position. Pairwise Wright’s FST distances between the Andean study population of interest, PEL, and CHB were calculated with PLINK.
Selection scan signals were identified by averaging the –log10(p-value) of iHS, as well as the XP-EHH and LSBL values, on a sliding window of 10 SNPs. We first evaluated selection scan signals common to all Andean study populations by annotating the top 0.5% of selected SNPs obtained with each positive selection method and population to genes using Variant Effect Predictor [32]. This 0.5% cut-off is a conservative approach compared to that used in other Andean studies in the literature, which used 1% or 5% cut-offs because their genotype data was more sparse than the data used in the current study [33,34]. We searched for genes that were within 10 kb on either side of the input SNP to investigate potential distant regulatory effects of SNPs. A gene list was obtained for each selection scan method (n = 3) and population of interest (n= 3), resulting in 9 gene lists in total. Genes overlapping across populations and selection scan methods were identified with GeneVenn [34]. We analyzed the overlap across populations based on genes instead of SNPs since we used the averaged selection scans to minimize single-SNP outliers.
We then evaluated traits that had undergone positive selection at a population level by selecting the top five selection regions for each population and method. From each of these regions, we selected the top SNP and identified the genes within 200 kb of either side of the SNP using Genome Browser.
We performed pathway enrichment analyses to determine the biological implications of the putatively selected genes shared across the three Andean study groups (Aymara-Quechua, Uru, and SAC). First, we used the Ingenuity Pathway Analysis software (content version 68752261; Ingenuity Systems, USA), using Fisher’s exact test. We did not correct for multiple comparisons in the IPA pathway analysis as the genes selected were based on 0.5% of SNPs obtained with each positive selection method and population, which is a conservative cut-off. Then, we carried out over-representation analyses (ORA) of the list of putatively selected genes across Andean study populations with the WebGestalt platform [35], using the KEGG and PANTHER Pathway databases. Here, we corrected for multiple testing to be stricter than the more explorative analysis by using IPA. WebGestalt uses the FDR method of Benjamini and Hochberg [36]. Pathway enrichment analysis is relevant when the study genes have cell signaling functions and therefore likely participate in several molecular and biological pathways. However, it does not necessarily pinpoint the selective pressure driving such positive selection sweeps in the genome.
We previously evaluated the population structure of the Bolivian and Argentinean study groups [9]. For this, we assessed the genetic affinities of the three Andean study groups in comparison to indigenous populations from North and South America [20,21], Europe, and Africa (to account for admixture). The Andean populations clustered together in principal component analysis (Figure S1A). Furthermore, in the population structure analysis, the ancestry fraction at K = 3 for European origin was 5.6% for SAC, 1.9% for Aymara-Quechua, and 0.2% for Uru, indicating limited European admixture in the three Andean study groups (Figure S1B).
Groups with a history of isolation often show high levels of homozygosity across their genomes. By assessing homozygosity for the three study groups with comparative data from the Americas (as assembled previously [21]; Figure S2), we found that the Andean study groups had levels of extended homozygosity comparable to other Native American populations, and in particular to groups that have limited non-Native American admixture. We also observed a low number of long homozygous regions (>8 Mb) in the Andean groups (including Uru), implying limited recent endogamy.
To study the extent and nature of the shared signals of positive selection between three populations from the Andes we used the lists of variants and genes by three selection tests reported in De Loma et al. 2022 study: two based on haplotype length (integrated haplotype score [iHS] and cross-population extended haplotype homozygosity [XP-EHH]) and one based on pairwise population differentiation at each given locus (locus-specific branch length [LSBL]). To minimize the detection of spurious single nucleotide polymorphism (SNP) outliers, we calculated the average p-values for iHS (Figure 1), and the average XP-EHH and LSBL values across the whole genome by sliding-window averaging (Figure S3 and Figure S4). For each selection scan method and population, we then focused on the top 0.5% candidate SNPs. Subsequently, we identified genes within 10 kb of either side from each top SNP. The number of putatively selected genes detected in the SAC, Aymara-Quechua, and Uru populations were 436, 507, and 454 for iHS; 340, 343, and 312 for XP-EHH; and 718, 720, and 882 for LSBL, respectively.

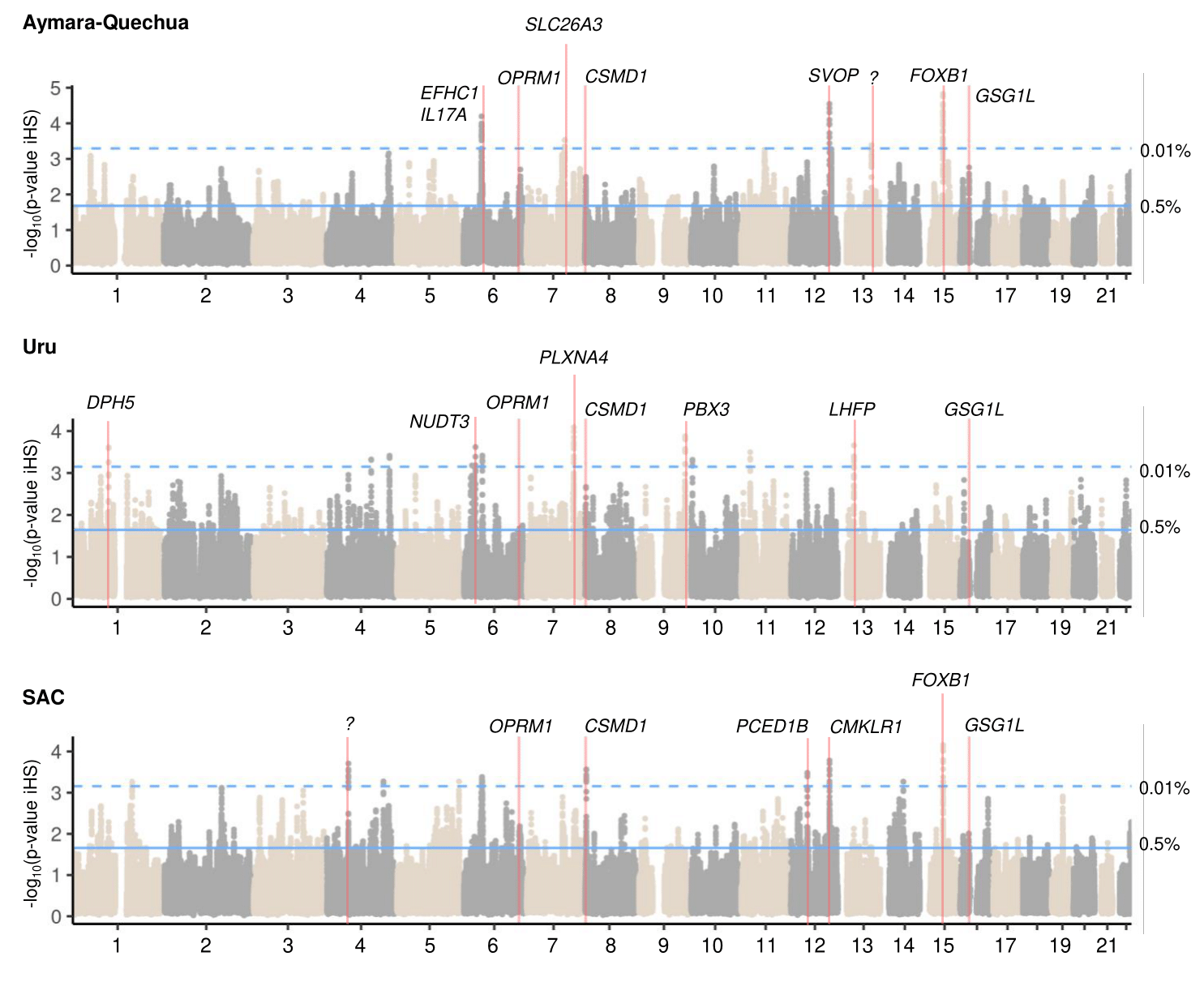
Figure 1. Genome-wide iHS selection scans for the three Andean study populations. To avoid spurious results due to single SNPs, we averaged the –log10(p-value) of iHS with a sliding window of 10 SNPs. The top five peaks in each population are marked with red lines, as well as the locations of OPRM1, CSMD1, and GSG1L. The top 0.5% (solid line) and top 0.01% (dashed line) are shown.
We evaluated adaptation-related traits common to all three Andean populations by identifying putatively selected genes (from any of the three selection scan methods) that overlapped across the three populations. This approach accounts for the fact that different selection scan methods are based on different principles, and therefore not all signals of positive selection are expected to be identified by all three methods. A total of 116 putatively selected genes were shared across all three Andean study groups (Figure 2A, Table S1). Among genes shared between two of the three groups, more genes were shared between the Aymara-Quechua and SAC populations (n = 307) than between either of those and the Uru population (n = 151 shared with SAC, n = 160 shared with Aymara-Quechua; Figure 2A, Table S1). For example, the arsenic methylating gene AS3MT was shared between the Aymara-Quechua and Uru populations, and the opioid receptor gene OPRM1 was shared between the SAC and Aymara-Quechua groups (Figure 3A).
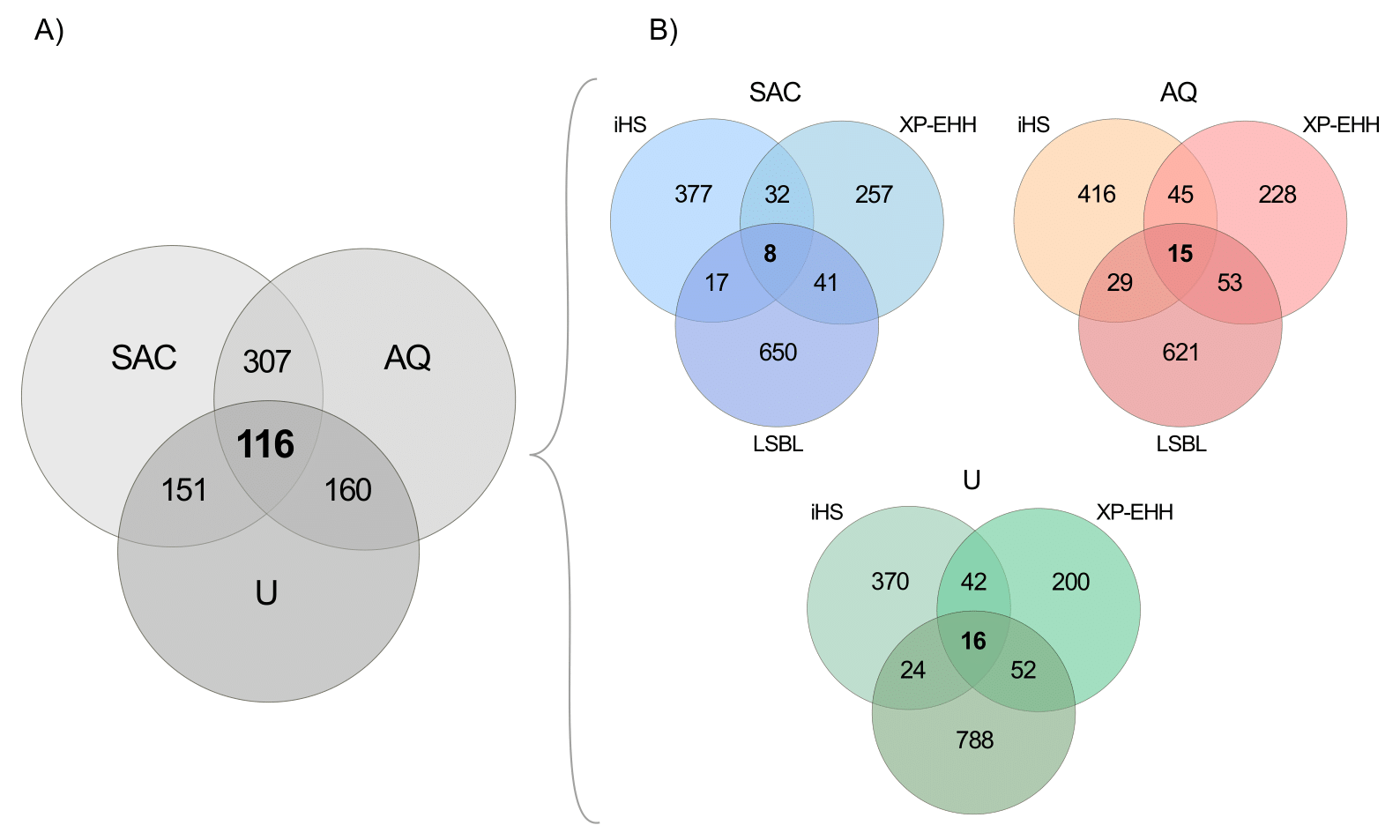
Figure 2. Genes potentially under positive selection identified by the three independent selection scan methods in the three Andean study populations (SAC, San Antonio de los Cobres; AQ, Aymara-Quechua; U, Uru). Genes included in the Venn diagrams are those annotated as being within ±10 kb from the top 0.5% SNPs identified by each selection scan method and in each population. (A) Venn diagram of putatively selected genes (identified by any of the selection scan methods) that are common among the three Andean study populations. (B) Venn diagrams per population of putatively selected genes shared across selection scan methods.
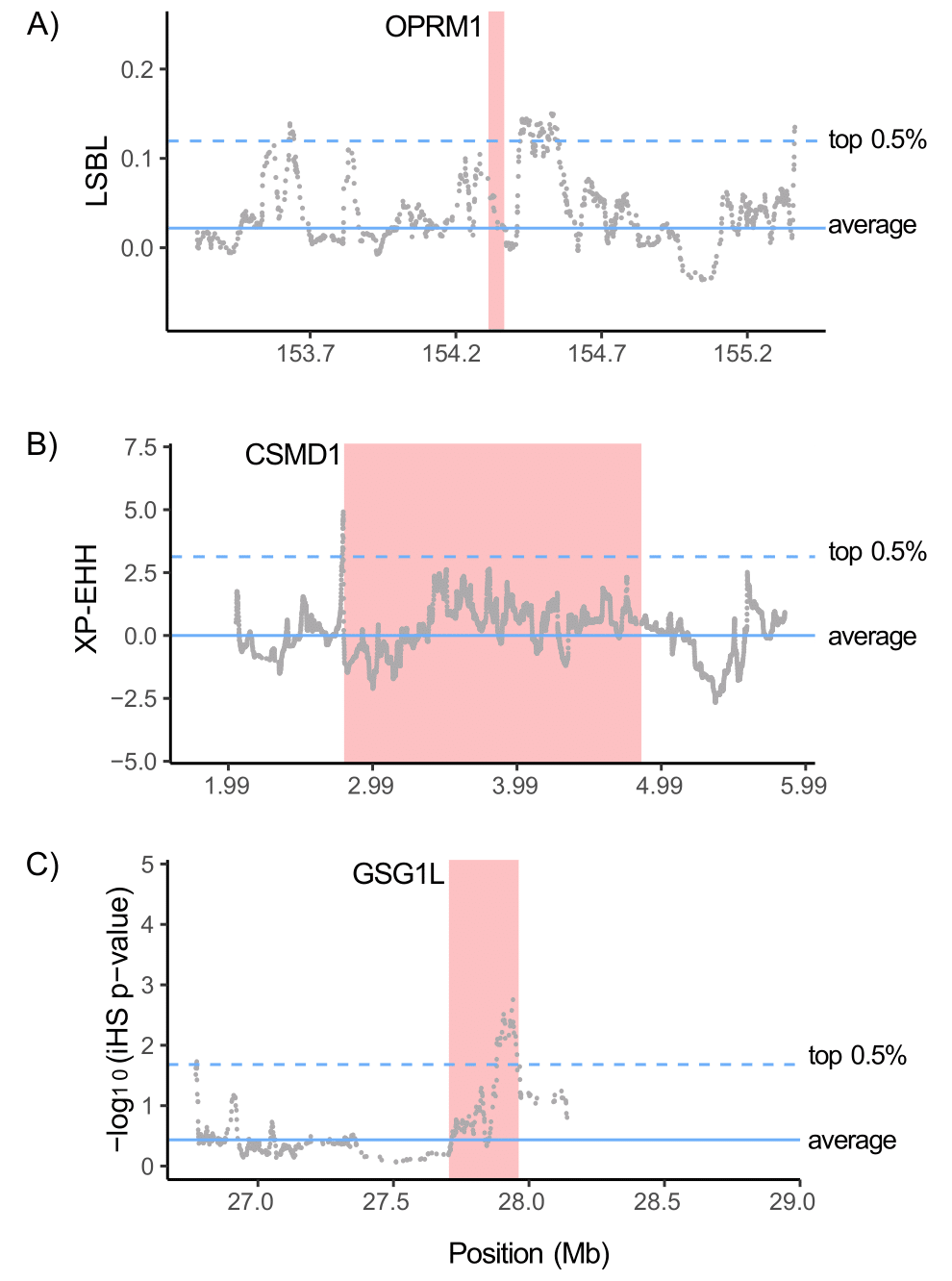
Figure 3. Zoom-ins of selection scans for OPR1M (chromosome 6), CSMD1 (chromosome 8), and GSG1L (chromosome 16) in the Aymara-Quechua study population (n = 65). The width of the red line indicates the size of each gene. The most representative selection scan method for each gene was included. The genome-wide average (solid line) and top 0.5% (dashed line) are shown.
To identify whether the putatively selected genes shared across the Andean study populations were over-represented in specific biological pathways and/or functions, we performed gene-enrichment pathway analyses (Table 1). The Ingenuity Pathway Analysis (IPA) software mapped 90 of the 116 shared gene IDs to specific loci in the IPA database; 26 gene IDs that encoded non-coding RNAs were not in the IPA database (Table S2). IPA canonical pathway analysis showed significant enrichment for the sperm motility pathway, as well as multiple pathways related to immune defense against pathogens, cardiovascular functions (including the renin-angiotensin pathway, although not statistically significant), and neurological signaling (Table 1, Table S3). In addition, there was an enrichment for the opioid signaling pathway (Table 1, Table S3). In the disease or function analysis, 71 out of 90 shared genes were associated with cancer (Table S4).
Table 1. Gene-enrichment analyses for putatively selected genes shared among Andean populations.
Putatively selected genes were those at ± 10 kb from the top 0.5% SNPs in each selection scan method. Enrichment pathway analyses were performed on the 116 genes that were shared among the Andean study populations according to any of the three selection scan methods. Two different programs were used: Ingenuity Pathway Analysis (IPA) and WebGestalt. The number of mapped genes in the datasets was 90 for IPA, 21 for KEGG, and 7 for PANTHER. Rank is within each dataset.
We performed complementary over-representation analyses using the web-based application WebGestalt and the KEGG and PANTHER Pathway databases. The morphine addiction and renin secretion pathways in KEGG, although not statistically significant after adjustment for false discovery rate (FDR, p-value < 0.05), were enriched among the shared putatively selected genes (Table 1, Table S5). In the PANTHER Pathway analysis, the histamine H1 receptor-mediated signaling pathway, related to inflammation, was enriched, along with the muscarinic acetylcholine receptor 1 and 2 signaling pathway (Table 1, Table S6). Furthermore, the endothelin signaling pathway was included within the top 10 results of the PANTHER Pathway analysis (Table S6).
In addition, we evaluated, within each population, whether any of the top positive selection signals (top 0.5% of SNPs) were shared across the three selection scan methods (Figure 2B). The number of putatively selected genes identified by all three selection scan methods were 8 for SAC (Table S7), 15 for Aymara-Quechua (Table S8), and 16 for Uru (Table S9). We then assessed whether any of these genes were also present in the list of 116 putatively selected genes shared across all three Andean populations (marked in bold in Tables S7–S9). The only protein-coding genes that were putatively selected in all three selection scan methods for more than one population were: CUB and Sushi Multiple Domains 1 (CSMD1), which encodes a protein involved in the regulation of the complement system among other functions [37], in the Aymara-Quechua and Uru populations; and Germ Cell-Specific Gene 1-Like (GSG1L), a membrane protein that regulates synaptic transmission in the brain [38], in the SAC and Aymara-Quechua groups (Figure 3B and Figure 3C). No protein-coding genes were putatively selected in all selection scan methods and in all three populations. However, Y RNA, a class of small non-coding RNAs, was putatively selected in all three populations and in all three methods. Furthermore, snoU13, a small nucleolar non-coding RNA, was selected in all three methods in the SAC and Uru study groups.
We next investigated the top five regions identified by each selection scan method in each population (Figure 1, Figure S3, and Figure S4 and Tables S10–S12). Since this is the approach most commonly used in adaptation studies in the Andes, where fewer methods and populations have been analyzed simultaneously, we included this approach to facilitate comparison with the literature deriving from this geographic area. Several genes in the top five selection regions were previously identified in selection studies on adaptation to high altitude, for example CPNE4, CSMD1, FOXB1, IL17F, and SP100, and in studies related to arsenic adaptation, such as AS3MT and LHFP (Tables S10–S12). In addition, several of the genes (n = 14, for example CPNE4, CSMD1, LHFP, SLC26A3, and TSPAN5) within the top selection peaks were present in the list of 116 putatively selected candidate genes shared across all Andean study populations (marked in bold in Tables S10–S12). Furthermore, two protamine genes (PRM1 and PRM2), potentially related to sperm motility, that scored highest in the IPA enrichment analysis, were the third highest XP-EHH signal in the SAC study group.
The Andes provides a unique setting for investigating human adaptation to extreme environments. In this study, we evaluated positive selection signals at a genome-wide level using three methods across three indigenous Andean populations, one from Argentina and two from Bolivia. Although not significant after adjustment for multiple testing, among the putatively selected genes, we identified genes involved in opioid signaling and morphine addiction as well as OPRM1, an opioid receptor. Other putatively selected genes were related to sperm motility, including the sperm DNA-packing protamine genes PRM1 and PRM2, and to high altitude adaptation (CSMD1 and ALDH51A). This study broadens our understanding of how humans adapted to the harsh environment of the Andes under multiple selection pressures. The positive selection of genes involved in the opioid signaling pathway is potentially related to how indigenous people adapted culturally (by routinely chewing coca leaves) to handle the extreme Andean conditions.
The three Andean groups had limited genetic influence from Europeans during the colonial period. In the case of the Uru study group, only 0.2% of their genome shows high similarity to European genomes, while the SAC and Aymara-Quechua groups show 5.6% and 1.9% similarity, respectively. The Uru have historically been isolated from neighboring communities [18]. Despite this isolation, we did not observe long stretches of homozygosity (> 8 Mb long) in the Uru genomes, which indicates that recent inbreeding has been limited. This is the first study to include Uru individuals in a genome-wide context, allowing us to expand our understanding of the peopling of the Andes. To date, this Andean community has been represented in the literature only through genetic analyses of Y-chromosome and mitochondrial DNA (mtDNA) data, which already pointed to their distinctive ancestry [39].
Within the putatively selected genes across all Andean study groups, we found signals near genes that participate in multiple signaling pathways, including sperm motility, immune defense against pathogens, and vascular functions. Although the overlap between signaling pathways and the few significant genes per pathway hinder identification of the most likely function(s) for these genes in relation to their selection pressure, all of these genes may be relevant for adaptation to living in the Andes. We discuss their different potential functions below. Furthermore, the identification of positive selection in genomic regions of non-coding RNAs needs further investigation. For example, snoU13 was identified in all three selection scan methods in the SAC and Uru populations. Although the function of snoU13 is still unclear, it was previously detected in positive selection scans in Tibetan domestic pigs adapted to high altitude [40].
We identified signals of positive selection around OPRM1 and other genes related to opioid signaling (ITPR2, KCNJ5, and PRKCH) and morphine addiction (KCNJ5, PDE3A, and PDE4D). In addition, we found enrichment for the muscarinic acetylcholine receptor signaling pathway, which is linked to the opioid signaling pathway [41]. The opioid signaling pathway and its receptors interact with drugs such as morphine, methadone, and cocaine, and influence dependence on these substances [42]. Rats given cocaine showed a dose-response induction of OPRM1 in the brain (Soderman and Unterwald 2009) [43]. Genetic variants of OPRM1 have been linked to cocaine, heroin, and alcohol consumption in populations of European ancestry [44,45]. Interestingly, genetic variants of GSG1L (the only putatively selected gene in all three selection scan methods in both Bolivian populations) are associated with the physiological response to methadone in heroin-dependent patients [46].
Chewing coca leaves, which contain cocaine, is a well-known and longstanding practice in the Andean region as part of rituals and to prevent or mitigate altitude sickness, hunger, fatigue, and pain [47]. Traces of cocaine in mummies from Chile suggest that this practice dates back at least 3,000 years [47]. Opioids and cocaine have a central role in regulating pain, and sequence variants of OPRM1 seem to modify how humans handle pain [48]. A study in Peru highlighted that children with altitude-induced chronic hypoxia, living in Cusco (3399 m above sea level) and mostly from Andean indigenous communities, required 40% less opioids for analgesia than those living in Lima at sea level [49]. In this context, our identification of putatively selected genes related to opioid signaling potentially implicates an ethnicity-specific mechanism of pain perception, driven by adaptation to the use of coca leaves to overcome altitude sickness, hunger, fatigue, and pain.
Selection of genes in opioid-related pathways have been reported before, but not related to adaption to a selection pressure, such as cope with living in the Andes. Rockman et al. (2005) found signals of selection at the human prodynorphin gene, which is an opioid polypeptide hormone [50].
At least four signaling pathways related to the immune system were significantly enriched within the 116 putatively selected genes common across the three Andean populations. Furthermore, our complementary enrichment analysis identified genes involved in the histamine H1 receptor signaling pathway. The histamine H1 receptor participates in the immune response, and its antagonists are regularly used against allergies. The histamine H1 receptor expression is modulated by Mycobacterium tuberculosis, which causes tuberculosis [51]. Recently, a study on indigenous people living in the Ecuadorian highlands showed positive selection for immune function related to tuberculosis [52]. Other antagonists of the histamine H1 receptor are effective against parasitic infections, such as leishmaniasis [53] and Chagas diseases [54].
Another gene that was putatively selected across all Andean populations and is involved in the defense against pathogens was Mucin 17 (MUC17). Mucins are involved in the immune response and host–parasite interactions [55]. Previous selection studies in humans also identified mucin genes, such as MUC19, in a population from central Mexico [56]. Positive selection of mucin genes could lead to resistance to infection by parasites and other pathogens.
The top pathway enriched in the IPA analysis was the sperm motility pathway. In extreme environmental conditions, such as in the Andes with high altitude, harsh temperatures, and water scarcity, reproductive fitness is crucial. This is reflected in the selection scans of the SAC study group, where we identified within the top selection peaks signals near PRM1 and PRM2, which encode protamines involved in the condensation of chromatin during sperm formation [57]. Protamine genes are rapidly evolving in humans because of positive selection [58,59]. Although the evolution of protamine genes has been widely studied across species, few have reported positive selection in human indigenous populations in relation to specific environmental factors. A study including the native American populations from the Human Origins project [21], identified signs of positive selection in the region including the protamine genes [60].
In addition, CSMD1 was identified through all selection scan methods in the Aymara-Quechua and Uru populations, as well as in the LSBL and iHS scans in the SAC group. Although its most characterized function is related to inflammation, rare mutations in CSMD1 have been linked to infertility in males and females [61]. Recently, higher reproductive fitness of native Tibetans at high altitude was linked to polygenic adaptation [62].
We previously identified signals of positive selection near AS3MT, which encodes the main methyltransferase enzyme that metabolizes arsenic in humans, linked to efficient arsenic metabolism in the study groups from the Argentinean Andes [10] and the Bolivian Andes [9]. In the current study, despite having fewer individuals in each study group, AS3MT was still within the top selection signal for the LSBL method in the Aymara-Quechua population (Table S12), and was shared between the Aymara-Quechua and Uru in all of the methods (Table S1). Since the first description of arsenic adaptation in humans [10], selection for AS3MT has been identified in study groups from Argentina, Chile, Peru, and Bolivia [9,63-65]. Together, these results suggest that there has been strong selective pressure for metabolizing arsenic in several groups across the Andean Mountain range. However, the causal variants driving this adaptation to arsenic have not been identified but a recent study suggests multiple potential causal variants, some ethnically distinct, around AS3MT that are influencing arsenic metabolism [66]. It should be stressed that even if these populations have a genetically determined efficient arsenic metabolism, they are not immune to arsenic toxicity, and further efforts should be made to lower arsenic exposure in these areas.
Current evidence implicates multiple genes in the adaptation to high altitude and indicates that these genes differ depending on the study population and the region [67]. In our study, we found two genes potentially associated with high-altitude adaptation in the Andes: CSMD1 and ALDH5A1 (Tables S10–S12 show all genes previously linked to altitude adaptation within the top selection peaks). CSMD1 participates in the complement pathway involved in inflammation, and it is considered a tumor suppressor whose allelic loss is associated with poor prognosis in epithelial cancers [37]. This gene has also been detected in selection scans for altitude adaptation in Tibetans, although it was not among the top candidates in that population [68]. Still, how it could confer adaptation to high altitude is unknown.
ALDH5A1, a candidate selected gene shared among all populations in our study, encodes an aldehyde dehydrogenase that catalyzes a step in the degradation of the neurotransmitter gamma-aminobutyric acid (GABA). Deficiency of this enzyme is known as 4-hydroxibutyricaciduria, a rare autosomal metabolic disease that leads to an abnormal accumulation of 4-hydroxibutyric acid (GHB) [69]. Under hypoxia, ischemia, or excessive metabolic demands, GHB levels rise in order to protect central and peripheral tissues [70,71]. Since GHB is considered to protect against hypoxic conditions, increased ALDH5A1 expression could be beneficial during altitude-induced hypoxia.
Several putatively selected genes were enriched in pathways related to renin-angiotensin signaling (ITPR2 and PRKCH) and renin secretion (ITPR2 and PDE3A), which could potentially be connected to high altitude adaptation. Changes in fluid retention and regulation of blood pressure are physiological processes involved in high altitude acclimatization. Indeed, genetic variants of the angiotensin-converting enzyme (ACE) have been associated with adaptation to high altitude [72]. Interestingly, adaptation to high altitude in the Andes is linked to increased hemoglobin levels in blood, compared to high-altitude Tibetan populations where this phenotype is not seen to the same extent [73]. High levels of hemoglobin in blood are a risk factor for cardiovascular disease [74,75]. Therefore, we speculate that potentially selected pathways related to cardiovascular functions may reflect a need to compensate for the higher hemoglobin levels induced to cope with hypoxia in the Andes at high altitude.
We did not find signal of selection around the HIF2A locus [7] or other genes previously linked to high altitude adaptation. It should be noted that some of the PEL population living at low altitude in Lima may be recent migrants from high altitude and therefore altitude adaptation signals may not be that strong. Nonetheless, in the present study we still identify several genes linked to altitude adaptation, suggesting that using PEL as the reference group provides adequate power to identify altitude adaptation.
The enrichment analysis identified 71 out of 90 putatively selected genes as associated with different types of cancer. A recent study of Native American ancestry and cancer mortality risk in Chile showed that a greater degree of Aymara descent is significantly associated with increased mortality due to skin, bladder, larynx, bronchus, and lung cancer [76]. Populations in the highlands of Ecuador also have an elevated incidence of several types of cancer, including colon, hematopoietic, and liver cancers, supporting the possibility of a higher cancer risk among people living at high altitude [77]. A recent metanalysis found that genes selected for adaptation to extreme environments, such as high altitude or cold, are enriched for cancer-associated genes [78]. These previously reported cancer patterns differ from the pattern of top cancer types we found in our enrichment analysis. However, when interpreting the findings for cancer and cancer profiles, it is worthwhile considering the current over-representation of cancer data in the literature and the existing bias for certain cancer forms. Alternatively, the results reflect that genetic variants selected to enhance survival under extreme conditions could be associated with pleiotropic effects including promoting cancer risk.
In conclusion, we identified novel selection signatures related to opioid signaling, sperm motility, and altitude adaptation common among Andean populations in the Bolivian Altiplano and the Argentinean Puna. There are a longstanding custom of chewing coca leaves in the Andes to mitigate altitude sickness, hunger, fatigue, and pain. Considering this, selection for opioid signaling could conceivably represent a mechanism for coping with stressors during human settlement in the harsh environment of the Andes. This potential connection between modified opioid signaling and coca-leaf chewing provides evidence of possible gene–cultural co-evolution in adaptation to high altitude. Our positive selection scans also found that the reproductive fitness of these populations, immune defense against pathogens, and high-altitude adaptation may have been driven by genetic factors.
The following supplementary materials are available on the website of this paper: HPGG2505010002SupplementaryMaterials.zip
Figure S1: Population structure of the three Andean study populations (marked in bold) and comparative populations.
Figure S2: Runs of homozygosity of the Andean study populations and comparative populations.
Figure S3: Genome-wide XP-EHH selection scans for the three Andean study populations.
Figure S4: Genome-wide LSBL selection scans for the three Andean study populations.
Table S1: Putatively selected genes overlapping across the Andean populations according to any selection scan method.
Table S2: Gene symbol mapping by IPA from the list of putatively selected genes shared across the three Andean populations (n = 116).
Table S3: List of pathways (IPA; Canonical) enriched in the putatively selected genes across the three Andean populations. Top 50 results.
Table S4: List of pathways (IPA; Disease or Function) enriched in the putatively selected genes across the three Andean populations. Top 50 results.
Table S5: List of pathways (WebGestalt; KEGG) enriched in the putatively selected genes across the three Andean populations. Top 10 results.
Table S6: List of pathways (WebGestalt; PANTHER) enriched in the putatively selected genes across the three Andean populations. Top 10 results.
Table S7: Putatively selected genes overlapping across selection scan methods in the SAC study population.
Table S8: Putatively selected genes overlapping across selection scan methods in the Aymara-Quechua study population.
Table S9: Putatively selected genes overlapping across selection scan methods in the Uru study population.
Table S10: Genes within the top five peaks for potential positive selection in the iHS selection scans.
Table S11: Genes within the top five peaks for potential positive selection in the XP-EHH selection scans.
Table S12: Genes within the top five peaks for potential positive selection in the LSBL selection scans.
Ethical approval for this study was obtained by the Comisión Provincial de Ética del Ministerio de Salud de Salta (Argentina), the Comité Nacional de Bioética (Bolivia), and the Regional Ethic Committee of Karolinska Institutet (Sweden, no. 2020-00495 and 2020-00493).
Informed consent for the publication of this study from the concerned individual has been obtained.
The genome-wide genotypes presented in this paper will be made available under controlled access via Federated European Genome-phenome Archive (FEGA) Sweden once this repository becomes operational. The datasets will then be findable through the European Genome-phenome Archive web portal (https://ega-archive.org). Until the dataset included in this study is available at SciLifeLab Data Repository (https://figshare.scilifelab.se/articles/dataset/_b_Human_adaptation_in_the_Andes_Mountains_b_/25323256)
This work was supported by the Institut de Recherche pour le Développement, Bolivia; the Hydrosciences Montpellier Laboratory, France; the Eric Philip Sörensens Stiftelse, Sweden; the Swedish Research Council, Sweden (2017-01239 and 2020-02827 to K. Broberg, and 2014-5211 to C. Schlebusch); Karolinska Institutet, Sweden; the Swedish International Development Cooperation Agency, Sweden (2016-05799 and 2019-03832 to K. Broberg); and the Genetics Institute at the Universidad Mayor de San Andrés, Bolivia.
Carina Schlebusch is a member of the Editorial Board of the journal Human Population Genetics and Genomics. The author was not involved in the journal’s review of or decisions related to this manuscript. The authors have declared that no other competing interests exist.
Conceptualization: K.B. and C.S.; Methodology: J.D.L., M.V. C.S., K.B.; Software: M.V.; Formal analysis: J.D.L and M.V.; Investigation: J.D.L. J.G. N.T. K.B., F.A. L.P.; Writing – Original Draft: J.D.L.; Writing – Review & Editing: J.D.L. J.G. N.T. K.B., F.A. L.P. C.S., M.V.; Visualisation: J.D.L., M.V.; Supervision: K.B., C.S.; Funding acquisition: K.B., C.S., J.G., N.T.
We warmly thank all the participants who took part in this study. We would also like to thank the Servicio Departamental de Salud de Oruro (SEDES) and the Salud Familiar Comunitaria e Intercultural (SAFCI) program for their contributions during the field activities in Bolivia. We also thank the fieldwork teams and the workers at local health centers in San Antonio de los Cobres and in the villages around Lake Poopó for assisting during recruitment. We also thank Gabriela Concha for her invaluable contribution during the recruitment in San Antonio de los Cobres. We thank Marta Toscano and Paola Zago for helping with the informed consents in San Antonio de Los Cobres. We acknowledge Anda Gliga’s help regarding the pathway enrichment analyses. We thank Diana Ekman at the National Bioinformatics Infrastructure Sweden at SciLifeLab for productive discussions about SNP-to-gene location. Also, we are thankful to Chiara Barbieri for sharing her data prior to publication.
The genotyping was performed by the SNP&SEQ Technology Platform in Uppsala (www.genotyping.se). The facility is part of the National Genomics Infrastructure supported by the Swedish Research Council for Infrastructures and Science for Life Laboratory, Sweden. The SNP&SEQ Technology Platform is also supported by the Knut and Alice Wallenberg Foundation. The computations were performed at the Swedish National Infrastructure for Computing (SNIC-UPPMAX; Projects SNIC 2018/8-107 and SNIC 2021/22-262).
| 1. | Rademaker K, Hodgins G, Moore K, Zarrillo S, Miller C, Bromley GR, et al. Paleoindian settlement of the high-altitude Peruvian Andes. Science. 2014;346(6208):466-469. [Google Scholar] [CrossRef] |
| 2. | Amaru R, Song J, Reading NS, Gordeuk VR, Prchal JT. What we know and what we do not know about evolutionary genetic adaptation to high altitude hypoxia in Andean Aymaras. Genes. 2023;14(3):640. [Google Scholar] [CrossRef] |
| 3. | Bigham A, Bauchet M, Pinto D, Mao X, Akey JM, Mei R, et al. Identifying signatures of natural selection in Tibetan and Andean populations using dense genome scan data. PLoS Genet. 2010;6(9):e1001116. [Google Scholar] [CrossRef] |
| 4. | Bigham AW, Mao X, Mei R, Brutsaert T, Wilson MJ, Julian CG, et al. Identifying positive selection candidate loci for high-altitude adaptation in Andean populations. Hum. Genet. 2009;4:1-12. [Google Scholar] [CrossRef] |
| 5. | Crawford JE, Amaru R, Song J, Julian CG, Racimo F, Cheng JY, et al. Natural selection on genes related to cardiovascular health in high-altitude adapted Andeans. Am J Hum Genet. 2017;101(5):752-67. [Google Scholar] [CrossRef] |
| 6. | Fehren-Schmitz L, Georges L. Ancient DNA reveals selection acting on genes associated with hypoxia response in pre-Columbian Peruvian Highlanders in the last 8500 years. Sci Rep. 2016;6(1):23485. [Google Scholar] [CrossRef] |
| 7. | Jorgensen K, Song D, Weinstein J, Garcia OA, Pearson LN, Inclán M, et al. High-altitude Andean H194R HIF2A allele is a hypomorphic allele. Mol Biol Evol. 2023;40(7):msad162. [Google Scholar] [CrossRef] |
| 8. | Valverde G, Zhou H, Lippold S, de Filippo C, Tang K, López Herráez D, et al. A novel candidate region for genetic adaptation to high altitude in Andean populations. PloS one. 2015;10(5):e0125444. [Google Scholar] [CrossRef] |
| 9. | De Loma J, Vicente M, Tirado N, Ascui F, Vahter M, Gardon J, et al. Human adaptation to arsenic in Bolivians living in the Andes. Chemosphere. 2022;301:134764. [Google Scholar] [CrossRef] |
| 10. | Schlebusch CM, Gattepaille LM, Engström K, Vahter M, Jakobsson M, Broberg K. Human adaptation to arsenic-rich environments. Mol Biol Evol. 2015;32(6):1544-1555. [Google Scholar] [CrossRef] |
| 11. | Arango-Isaza E, Capodiferro MR, Aninao MJ, Babiker H, Aeschbacher S, Achilli A, et al. The genetic history of the Southern Andes from present-day Mapuche ancestry. Curr Biol. 2023;33(13):2602-15.e5. [Google Scholar] [CrossRef] |
| 12. | De Loma J, Tirado N, Ascui F, Levi M, Vahter M, Broberg K, et al. Elevated arsenic exposure and efficient arsenic metabolism in indigenous women around Lake Poopó, Bolivia. Sci Total Environ. 2019;657:179-186. [Google Scholar] [CrossRef] |
| 13. | Harari F, Langeén M, Casimiro E, Bottai M, Palm B, Nordqvist H, et al. Environmental exposure to lithium during pregnancy and fetal size: a longitudinal study in the Argentinean Andes. Environ Int. 2015;77:48-54. [Google Scholar] [CrossRef] |
| 14. | Núñez L, Hill HG, Martínez AL. Guia : Museo arqueológico Universidad Católica del Norte, Chile. San Pedro de Atacama, Chile: Universidad Católica del Norte; 1995. [Google Scholar] |
| 15. | Normando Cruz E. Historia y Etnicidad en las Yungas de la Argentina. Venezuela: Ediciones Polar; 2011. [Google Scholar] |
| 16. | Batai K, Williams SR. Mitochondrial variation among the Aymara and the signatures of population expansion in the central Andes. Am J Hum Biol. 2014;26(3):321-330. [Google Scholar] [CrossRef] |
| 17. | Lindo J, Haas R, Hofman C, Apata M, Moraga M, Verdugo RA, et al. The genetic prehistory of the Andean highlands 7000 years BP though European contact. Sci Adv. 2018;4(11):eaau4921. [Google Scholar] [CrossRef] |
| 18. | De la Barra Saavedra SZ, Barrientos ML, Cruz ROC. Exclusión y subalternidad de los urus del lago Poopó: Discriminación en la relación mayorías y minorías étnicas: Programa de Investigación Estratégica en Bolivia. 2011. |
| 19. | Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4(1):s13742-015-0047-8. [Google Scholar] [CrossRef] |
| 20. | Barbieri C, Barquera R, Arias L, Sandoval JR, Acosta O, Zurita C, et al. The current genomic landscape of western south America: Andes, amazonia, and Pacific coast. Mol Biol Evol. 2019;36(12):2698-713. [Google Scholar] [CrossRef] |
| 21. | Lazaridis I, Patterson N, Mittnik A, Renaud G, Mallick S, Kirsanow K, et al. Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature. 2014;513(7518):409-413. [Google Scholar] [CrossRef] |
| 22. | Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen W-M. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26(22):2867-2873. [Google Scholar] [CrossRef] |
| 23. | Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2(12):e190. [Google Scholar] [CrossRef] |
| 24. | Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904-909. [Google Scholar] [CrossRef] |
| 25. | Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19(9):1655-1664. [Google Scholar] [CrossRef] |
| 26. | Behr AA, Liu KZ, Liu-Fang G, Nakka P, Ramachandran S. Pong: fast analysis and visualization of latent clusters in population genetic data. Bioinformatics. 2016;32(18):2817-2823. [Google Scholar] [CrossRef] |
| 27. | Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4(3):e72. [Google Scholar] [CrossRef] |
| 28. | Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C, et al. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449(7164):913-918. [Google Scholar] [CrossRef] |
| 29. | Sabeti PC, Reich DE, Higgins JM, Levine HZ, Richter DJ, Schaffner SF, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419(6909):832-837. [Google Scholar] [CrossRef] |
| 30. | Gautier M, Vitalis R. rehh: an R package to detect footprints of selection in genome-wide SNP data from haplotype structure. Bioinformatics. 2012;28(8):1176-1177. [Google Scholar] [CrossRef] |
| 31. | Shriver MD, Kennedy GC, Parra EJ, Lawson HA, Sonpar V, Huang J, et al. The genomic distribution of population substructure in four populations using 8,525 autosomal SNPs. Hum. Genet. 2004;1:1-13. [Google Scholar] [CrossRef] |
| 32. | McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, et al. The ensembl variant effect predictor. Genome Biol. 2016;17:1-14. [Google Scholar] [CrossRef] |
| 33. | Eichstaedt CA, Antao T, Cardona A, Pagani L, Kivisild T, Mormina M. Genetic and phenotypic differentiation of an Andean intermediate altitude population. Physiol Rep. 2015;3(5):e12376. [Google Scholar] [CrossRef] |
| 34. | Pirooznia M, Nagarajan V, Deng Y. GeneVenn-A web application for comparing gene lists using Venn diagrams. Bioinformation. 2007;1(10):420. [Google Scholar] [CrossRef] |
| 35. | Wang J, Duncan D, Shi Z, Zhang B. WEB-based gene set analysis toolkit (WebGestalt): update 2013. Nucleic Acids Res. 2013;41(W1):W77-W83. [Google Scholar] [CrossRef] |
| 36. | Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Stat Method. 1995;57(1):289-300. [Google Scholar] [CrossRef] |
| 37. | Ma C, Quesnelle KM, Sparano A, Rao S, Park MS, Cohen MA, et al. Characterization CSMD1 in a large set of primary lung, head and neck, breast and skin cancer tissues. Cancer Biol. Ther. 2009;8(10):907-916. [Google Scholar] [CrossRef] |
| 38. | Gu X, Mao X, Lussier MP, Hutchison MA, Zhou L, Hamra FK, et al. GSG1L suppresses AMPA receptor-mediated synaptic transmission and uniquely modulates AMPA receptor kinetics in hippocampal neurons. Nat Commun. 2016;7(1):10873. [Google Scholar] [CrossRef] |
| 39. | Sandoval JR, Lacerda DR, Jota MS, Salazar-Granara A, Vieira PPR, Acosta O, et al. The genetic history of indigenous populations of the Peruvian and Bolivian Altiplano: the legacy of the Uros. PLoS One. 2013;8(9):e73006. [Google Scholar] [CrossRef] |
| 40. | Dong K, Yao N, Pu Y, He X, Zhao Q, Luan Y, et al. Genomic scan reveals loci under altitude adaptation in Tibetan and Dahe pigs. PLoS One. 2014;9(10):e110520. [Google Scholar] [CrossRef] |
| 41. | Margas W, Mahmoud S, Ruiz-Velasco V. Muscarinic acetylcholine receptor modulation of mu (μ) opioid receptors in adult rat sphenopalatine ganglion neurons. J Neurophysiol. 2010;103(1):172-182. [Google Scholar] [CrossRef] |
| 42. | Unterwald EM. Regulation of opioid receptors by cocaine. Ann N Y Acad Sci. 2001;937(1):74-92. [Google Scholar] [CrossRef] |
| 43. | Soderman AR, Unterwald EM. Cocaine-induced mu opioid receptor occupancy within the striatum is mediated by dopamine D2 receptors. Brain Res. 2009;1296:63-71. [Google Scholar] [CrossRef] |
| 44. | Zhang H, Luo X, Kranzler HR, Lappalainen J, Yang B-Z, Krupitsky E, et al. Association between two μ-opioid receptor gene (OPRM1) haplotype blocks and drug or alcohol dependence. Hum Mol Genet. 2006;15(6):807-819. [Google Scholar] [CrossRef] |
| 45. | Clarke T-K, Crist RC, Kampman KM, Dackis CA, Pettinati HM, O’Brien CP, et al. Low frequency genetic variants in the μ-opioid receptor (OPRM1) affect risk for addiction to heroin and cocaine. Neurosci Lett. 2013;542:71-75. [Google Scholar] [CrossRef] |
| 46. | Yang H-C, Chu S-K, Huang C-L, Kuo H-W, Wang S-C, Liu S-W, et al. Genome-wide pharmacogenomic study on methadone maintenance treatment identifies SNP rs17180299 and multiple haplotypes on CYP2B6, SPON1, and GSG1L associated with plasma concentrations of methadone R-and S-enantiomers in heroin-dependent patients. PLoS Genet. 2016;12(3):e1005910. [Google Scholar] [CrossRef] |
| 47. | Rivera MA, Aufderheide AC, Cartmell LW, Torres CM, Langsjoen O. Antiquity of coca-leaf chewing in the south central Andes: a 3,000 year archaeological record of coca-leaf chewing from northern Chile. J Psychoactive Drugs . 2005;37(4):455-458. [Google Scholar] [CrossRef] |
| 48. | Peciña M, Love T, Stohler CS, Goldman D, Zubieta J-K. Effects of the Mu opioid receptor polymorphism (OPRM1 A118G) on pain regulation, placebo effects and associated personality trait measures. Neuropsychopharmacology. 2015;40(4):957-965. [Google Scholar] [CrossRef] |
| 49. | Rabbitts JA, Groenewald CB, Dietz NM, Morales C, Raesaenen J. Perioperative opioid requirements are decreased in hypoxic children living at altitude. Paediatr Anaesth. 2010;20(12):1078-1083. [Google Scholar] [CrossRef] |
| 50. | Rockman MV, Hahn MW, Soranzo N, Zimprich F, Goldstein DB, Wray GA. Ancient and recent positive selection transformed opioid cis-regulation in humans. PLoS Biol. 2005;3(12):e387. [Google Scholar] [CrossRef] |
| 51. | Mo S, Guo J, Ye T, Zhang X, Zeng J, Xu Y, et al. Mycobacterium tuberculosis utilizes host histamine receptor H1 to modulate reactive oxygen species production and phagosome maturation via the p38MAPK-NOX2 axis. MBio. 2022;13(5):e02004-22. [Google Scholar] [CrossRef] |
| 52. | Joseph SK, Migliore NR, Olivieri A, Torroni A, Owings AC, DeGiorgio M, et al. Genomic evidence for adaptation to tuberculosis in the Andes before European contact. iScience. 2023;26(2). [Google Scholar] [CrossRef] |
| 53. | Pinto EG, da Costa-Silva TA, Tempone AG. Histamine H1-receptor antagonists against Leishmania (L.) infantum: an in vitro and in vivo evaluation using phosphatidylserine-liposomes. Acta Trop. 2014;137:206-210. [Google Scholar] [CrossRef] |
| 54. | De Rycker M, Thomas J, Riley J, Brough SJ, Miles TJ, Gray DW. Identification of trypanocidal activity for known clinical compounds using a new Trypanosoma cruzi hit-discovery screening cascade. PLoS Negl Trop Dis. 2016;10(4):e0004584. [Google Scholar] [CrossRef] |
| 55. | Hicks S, Theodoropoulos G, Carrington S, Corfield A. The role of mucins in host–parasite interactions. Part I–protozoan parasites. Parasitol Today. 2000;16(11):476-481. [Google Scholar] [CrossRef] |
| 56. | Reynolds AW, Mata-Míguez J, Miró-Herrans A, Briggs-Cloud M, Sylestine A, Barajas-Olmos F, et al. Comparing signals of natural selection between three Indigenous North American populations. Proc Natl Acad Sci U S A. 2019;116(19):9312-9317. [Google Scholar] [CrossRef] |
| 57. | Balhorn R. The protamine family of sperm nuclear proteins. Genome Biol. 2007;8:1-8. [Google Scholar] [CrossRef] |
| 58. | Wyckoff GJ, Wang W, Wu C-I. Rapid evolution of male reproductive genes in the descent of man. Nature. 2000;403(6767):304-309. [Google Scholar] [CrossRef] |
| 59. | Rooney AP, Zhang J. Rapid evolution of a primate sperm protein: relaxation of functional constraint or positive Darwinian selection? Mol Biol Evol. 1999;16(5):706-710. [Google Scholar] [CrossRef] |
| 60. | Refoyo-Martínez A, da Fonseca RR, Halldórsdóttir K, Árnason E, Mailund T, Racimo F. Identifying loci under positive selection in complex population histories. Genome Res. 2019;29(9):1506-1520. [Google Scholar] [CrossRef] |
| 61. | Lee AS, Rusch J, Lima AC, Usmani A, Huang N, Lepamets M, et al. Rare mutations in the complement regulatory gene CSMD1 are associated with male and female infertility. Nat Commun. 2019;10(1):4626. [Google Scholar] [CrossRef] |
| 62. | He Y, Guo Y, Zheng W, Yue T, Zhang H, Wang B, et al. Polygenic adaptation leads to a higher reproductive fitness of native Tibetans at high altitude. Curr Biol. 2023;33(19):4037-51.e5. [Google Scholar] [CrossRef] |
| 63. | Apata M, Arriaza B, Llop E, Moraga M. Human adaptation to arsenic in Andean populations of the Atacama Desert. Am J Phys Anthropol. 2017;163(1):192-199. [Google Scholar] [CrossRef] |
| 64. | Eichstaedt CA, Antao T, Cardona A, Pagani L, Kivisild T, Mormina M. Positive selection of AS3MT to arsenic water in Andean populations. Mutat Res. 2015;780:97-102. [Google Scholar] [CrossRef] |
| 65. | Jacovas VC, Couto-Silva CM, Nunes K, Lemes RB, de Oliveira MZ, Salzano FM, et al. Selection scan reveals three new loci related to high altitude adaptation in Native Andeans. Sci Rep. 2018;8(1):12733. [Google Scholar] [CrossRef] |
| 66. | Chernoff MB, Delgado D, Tong L, Chen L, Oliva M, Tamayo LI, et al. Sequencing-based fine-mapping and in silico functional characterization of the 10q24.32 arsenic metabolism efficiency locus across multiple arsenic-exposed populations. PLoS Genet. 2023;19(1):e1010588. [Google Scholar] [CrossRef] |
| 67. | Moore LG. Human genetic adaptation to high altitudes: current status and future prospects. Quat Int. 2017;461:4-13. [Google Scholar] [CrossRef] |
| 68. | Alkorta-Aranburu G, Beall CM, Witonsky DB, Gebremedhin A, Pritchard JK, Di Rienzo A. The genetic architecture of adaptations to high altitude in Ethiopia. PLoS Genet. 2012;8(12):e1003110. [Google Scholar] [CrossRef] |
| 69. | Lin C-Y, Weng W-C, Lee W-T. A novel mutation of ALDH5A1 gene associated with succinic semialdehyde dehydrogenase deficiency. J Child Neurol. 2015;30(4):486-489. [Google Scholar] [CrossRef] |
| 70. | Kamal RM, van Noorden MS, Franzek E, Dijkstra BA, Loonen AJ, De Jong CA. The neurobiological mechanisms of gamma-hydroxybutyrate dependence and withdrawal and their clinical relevance: a review. Neuropsychobiology. 2016;73(2):65-80. [Google Scholar] [CrossRef] |
| 71. | Vergoni AV, Ottani A, Botticelli AR, Zaffe D, Guano L, Loche A, et al. Neuroprotective effect of γ-hydroxybutyrate in transient global cerebral ischemia in the rat. Eur J Pharmacol. 2000;397(1):75-84. [Google Scholar] [CrossRef] |
| 72. | Wang Y, Lu H, Chen Y, Luo Y. The association of angiotensin-converting enzyme gene insertion/deletion polymorphisms with adaptation to high altitude: a meta-analysis. J Renin-Angiotensin-Aldosterone Syst. 2016;17(1):1470320315627410. [Google Scholar] [CrossRef] |
| 73. | Beall CM. Two routes to functional adaptation: Tibetan and Andean high-altitude natives. Proc Natl Acad Sci U S A. 2007;104 suppl_1:8655-8660. [Google Scholar] [CrossRef] |
| 74. | Lee G, Choi S, Kim K, Yun JM, Son JS, Jeong SM, et al. Association of hemoglobin concentration and its change with cardiovascular and all-cause mortality. J Am Heart Assoc. 2018;7(3):e007723. [Google Scholar] [CrossRef] |
| 75. | Corante N, Anza-Ramírez C, Figueroa-Mujica R, Macarlupú JL, Vizcardo-Galindo G, Bilo G, et al. Excessive erythrocytosis and cardiovascular risk in andean highlanders. High Alt Med Biol. 2018;19(3):221-231. [Google Scholar] [CrossRef] |
| 76. | Lorenzo Bermejo J, Boekstegers F, González Silos R, Marcelain K, Baez Benavides P, Barahona Ponce C, et al. Subtypes of Native American ancestry and leading causes of death: Mapuche ancestry-specific associations with gallbladder cancer risk in Chile. PLoS Genet. 2017;13(5):e1006756. [Google Scholar] [CrossRef] |
| 77. | Garrido DI, Garrido SM. Cancer risk associated with living at high altitude in Ecuadorian population from 2005 to 2014. Clujul Med. 2018;91(2):188. [Google Scholar] [CrossRef] |
| 78. | Voskarides K. Combination of 247 genome-wide association studies reveals high cancer risk as a result of evolutionary adaptation. Mol Biol Evol. 2018;35(2):473-485. [Google Scholar] [CrossRef] |

![]()
Copyright © 2026 Pivot Science Publications Corp. - unless otherwise stated | Terms and Conditions | Privacy Policy




