
Human Population Genetics and Genomics ISSN 2770-5005
Human Population Genetics and Genomics 2026;6(2):0006 | https://doi.org/10.47248/hpgg2606020006
Technical Note Open Access
KAlignedoscope: An interactive visualization tool for aligned clustering results from population structure analyses
Avery Guo
1,2
 ,
Sohini Ramachandran
3,4,†
,
Sohini Ramachandran
3,4,†
 ,
Xiran Liu
3,†
,
Xiran Liu
3,†
Correspondence: Sohini Ramachandran; Xiran Liu
Academic Editor(s): Joshua Akey, Carina Schlebusch, Torsten Günther
Received: Oct 2, 2025 | Accepted: Mar 10, 2026 | Published: Mar 27, 2026
© 2026 by the author(s). This is an Open Access article distributed under the Creative Commons License Attribution 4.0 International (CC BY 4.0) license, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly credited.
Cite this article: Guo A, Ramachandran S, Liu X. KAlignedoscope: An interactive visualization tool for aligned clustering results from population structure analyses. Hum Popul Genet Genom. 2026;6(2):0006. https://doi.org/10.47248/hpgg2606020006
Visualization plays an important role in the interpretation of analyses applied to population-genetic data, particularly when multiple clustering results are generated from the same input data and aligned to provide a comprehensive view of inferred population structure. We present KAlignedoscope, a web-based tool for the interactive visualization and exploration of aligned clustering results. Built with D3.js, our tool enables fast, dynamic rendering and offers powerful interactive features such as reordering populations and clusters, sorting individuals, highlighting clusters, and customizing colors. The tool is compatible with outputs from clustering alignment methods Clumppling and Pong, and is easily extendable to others. KAlignedoscope supports and streamlines population structure analysis by enabling flexible navigation of complex patterns in the aligned clustering results.
Keywordspopulation structure, clustering, clustering alignment, visualization, interactive
A central goal in population genetics is to infer ancestry and population structure from multilocus genotype data for a sample of individuals. Researchers typically apply model-based clustering (e.g.,
Clustering runs often produce substantially different solutions from run to run due to factors like algorithmic stochasticity, label permutation, multiple optima, and varying algorithm settings (e.g., number of clusters, K). To jointly analyze and interpret results from multiple clustering runs of population structure inference, several clustering alignment methods have been developed [4–7]. Clustering alignment methods typically work by grouping runs with the same K value into “modes”, which represent distinct clustering solutions (sometimes with varying levels of support) for the genetic data [4–7]. For any given K, the mode to which most clustering runs align, i.e., the most frequent solution, is referred to as the “major mode”. Other “minor modes” often reveal alternative patterns that become clearer at higher values of K. Modes are then aligned across different K values to show how clusters emerge.
Most clustering alignment methods display aligned modes as a vertical column of structure plots, with individuals aligned along the x-axis [4–6]. The most recent method,
Our D3.js-based visualization tool
Table 1. Interactive features in

In addition to the main features listed in Table 1,
Upon downloading,
Our tool is available as a light-weight Python package,
Figure 1 demonstrates the usage of our tool on aligned clustering results generated by
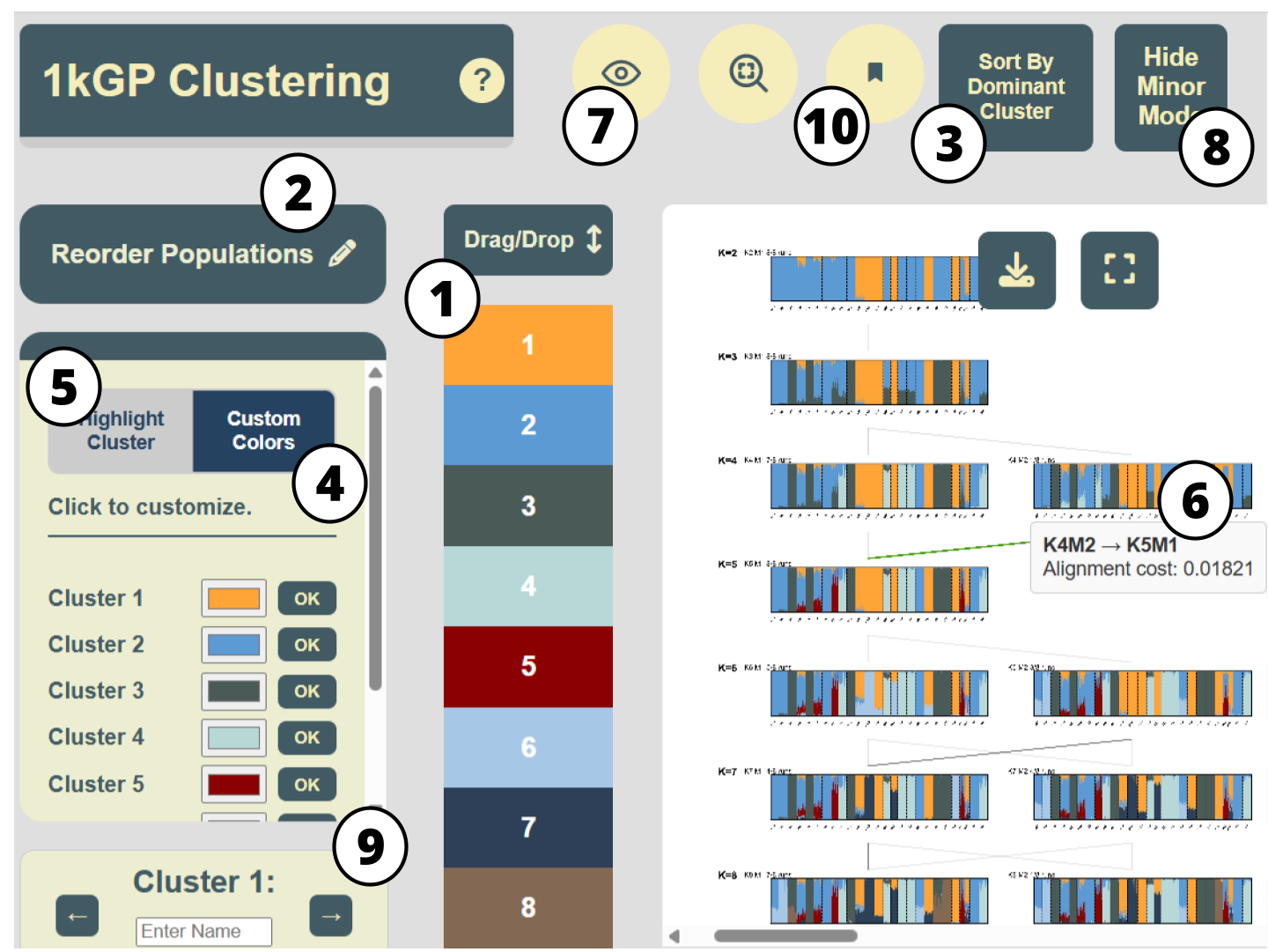
Figure 1. The
Taking in the membership matrices of the aligned modes as well as the alignment patterns (both are output of the
We focus on the structure plot of “K5M1” (the major mode for K = 5 runs) in Figure 2 to further demonstrate our interactive features. The initial rendering shown in Figure 2A uses the default ordering of individuals from the data. As a result, the clusters appear mixed and scattered, making it difficult to see the visual connections between them.
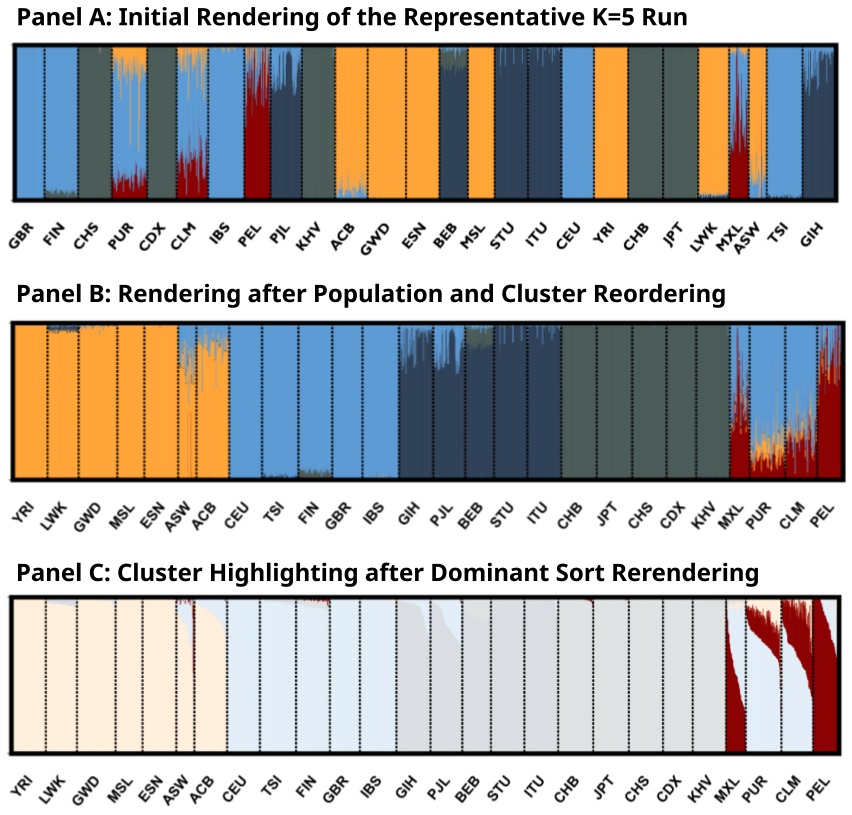
Figure 2. Zoomed-in structure plot of mode “K5M1” in Figure 1. (A) The default rendering of the clustering mode “K5M1” from
Highlighting A Cluster (Feature 5 in Table 1) helps focus on a specific aspect of the results. For example, in Figure 2C, a single highlighted cluster is clearly most common among several Admixed American populations (PUR, MXL, PEL, and CLM) from the 1kG data, suggesting both shared admixture proportion and variation in this shared proportion among these groups. Sorting by Cluster Dominance (Feature 3 in Table 1) rearranges the individuals into descending orders of each population’s largest cluster, which creates a cleaner and more intuitive view of how clusters are distributed. For example, the highlighted cluster in Figure 2C is a minor component for populations PUR (Puerto Ricans in Puerto Rico) and CLM (Colombians from Medellín, Colombia), but is dominating the population PEL (Peruvians from Lima). Each interactive feature is useful for examining different aspects of the data. We encourage users to explore and experiment to find the visual options that best fit their specific needs.
We developed
Visualization is not only a communication tool but also key for interpreting and evaluating population structure. Paired with population structure inference and clustering alignment methods, our tool streamlines population structure analysis workflows and provides comprehensive visualizations for exploring genetic admixture patterns.
Not applicable.
Data presented in the study are included in the GitHub repository of the tool at https://github.com/ramachandran-lab/KAlignedoscope.
This study was funded by the US National Institutes of Health (NIH R35 GM139628) to S.R. and the Brown Advanced Undergraduate Research Fellowship to A.G..
The authors have declared that no competing interests exist.
Conceptualization: S.R. and X.L.; Methodology: S.R. and X.L.; Software: A.G.; Data Curation: A.G., S.R., X.L.; Writing – Original Draft: A.G. and X.L.; Writing – Review & Editing: A.G., S.R., X.L.; Visualization: A.G.; Supervision: X.L.; Funding Acquisition: S.R.
We thank Xavier Roca Rada for testing the beta version of the tool and for providing valuable feedback that contributed to its improvement.
| 1. | Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945-959. [Google Scholar] [CrossRef] |
| 2. | Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19(9):1655-1664. [Google Scholar] [CrossRef] |
| 3. | Rosenberg NA. DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes. 2004;4(1):137-138. [Google Scholar] [CrossRef] |
| 4. | Jakobsson M, Rosenberg NA. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23(14):1801-1806. [Google Scholar] [CrossRef] |
| 5. |
Kopelman NM, Mayzel J, Jakobsson M, et al. Clumpak: a program for identifying clustering modes and packaging population structure inferences across |
| 6. | Behr AA, Liu KZ, Liu-Fang G, Nakka P, Ramachandran S. Pong: Fast analysis and visualization of latent clusters in population genetic data. Bioinformatics. 2016;32(18):2817-2823. [Google Scholar] [CrossRef] |
| 7. | Liu X, Kopelman NM, Rosenberg NA. Clumppling: cluster matching and permutation program with integer linear programming. Bioinformatics. 2024;40(1):btad751. [Google Scholar] [CrossRef] |
| 8. | Liu X, Rosenberg NA, Ramachandran S. Clumppling 2.0; 2025. Clumppling 2.0: A Clustering Alignment Program for Population Structure Analyses. Hum Popul Genet Genom. 2026;6(2):0004. [Google Scholar] [CrossRef] |
| 9. | Bostock M, Ogievetsky V, Heer J. D3 data-driven documents. IEEE Trans Vis Comput Graph. 2011;17(12):2301-2309. [Google Scholar] [CrossRef] |
| 10. | 1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68-74. [Google Scholar] [CrossRef] |

![]()
Copyright © 2026 Pivot Science Publications Corp. - unless otherwise stated | Terms and Conditions | Privacy Policy




