
Human Population Genetics and Genomics ISSN 2770-5005
Human Population Genetics and Genomics 2026;6(2):0007 | https://doi.org/10.47248/hpgg2606020007
Original Research Open Access
The reliability of inferred archaic segments
Nancy Bird

 ,
Erin Walker
,
Garrett Hellenthal
,
Erin Walker
,
Garrett Hellenthal
Correspondence: Nancy Bird; Garrett Hellenthal
Academic Editor(s): Joshua Akey
Received: Aug 31, 2025 | Accepted: Apr 27, 2026 | Published: May 15, 2026
© 2026 by the author(s). This is an Open Access article distributed under the Creative Commons License Attribution 4.0 International (CC BY 4.0) license, which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is correctly credited.
Cite this article: Bird N, Walker E, Hellenthal G. The reliability of inferred archaic segments. Hum Popul Genet Genom. 2026;6(2):0007. https://doi.org/10.47248/hpgg2606020007
Many or all present-day human genomes carry segments of DNA inherited from archaic humans due to admixture events occurring >25,000 years ago. Several methods have been published to detect such segments. These variously require phased haplotypes, an archaic reference sequence and/or an outgroup with little related archaic introgression. Inference from these approaches have been used to document purifying and positive selection of archaic segments and to understand their influence on the phenotype. However, the comparative accuracy of different methods at detecting archaic segments, and how well inferred segments overlap across methods, remains underexplored. Here, we used demographic simulations to evaluate accuracy in detecting archaic ancestry under three widely-used approaches: SPrime, IBDMix and HMmix, and a technique introduced here, cp-archaic, as well as applying all approaches to 1000 Genomes data. Our results reveal substantial variation in method performance, with recall and precision ranges differing significantly across approaches and parameter settings. cp-archaic achieved the highest accuracy overall (F1, the harmonic mean of precision and recall=0.92-0.94 across three different simulations). The choice of demographic simulation substantially impacted the distribution and characteristics of true archaic segments, with demographic scenarios creating both archaic ancestry ‘deserts’ and regions of very high archaic ancestry in the absence of selection. Notably, we also found low agreement between methods, with <22% overlap in individual archaic sites detected across all four approaches in real data. These findings highlight how conclusions about archaic introgression patterns, population differences, and genome-wide coverage depend critically on both methodological choice and underlying demographic assumptions. We recommend careful method selection and parameter optimisation, as well as caution when interpreting individual archaic segments, particularly in comparative studies across populations.
Keywordsarchaic introgression, Neanderthal, methods, local ancestry inference
It is generally accepted that many or all present-day human genomes contain segments of archaic (specifically Neanderthal and Denisovan-related) ancestry inherited from admixture events occurring over 25,000 years ago [1–7]. Several different methods have been published to infer archaic segments. These can rely on using reference data from a non-admixed outgroup, often an African population assumed not to have significant amounts of archaic ancestry, and/or archaic genomes, i.e., currently available high coverage Altai, Chagryskaya and Vindija Neanderthal genomes and/or one Altai Denisovan genome [1,8–10]. One method, IBDmix, does not require a non-admixed outgroup to run [11,12] and therefore can infer Neanderthal ancestry in African populations, which might be missed in methods that require an outgroup. There are several methods, such as SPrime and HMmix which do not require an archaic reference genome, but for which results can later be matched to available references [13–18]. Some methods, including cp-archaic introduced here, require both an outgroup and archaic reference [4,19,20].
The sets of archaic tracts inferred from these methods have been used to examine both the number of admixture pulses and the number of different archaic populations who introgressed into modern humans. The latter can be achieved by analysing the match rate of inferred archaic segments to different available archaic genomes. For example, evidence suggests that there were two or more different admixture events with distinct Denisovan populations in Asia [5,13,21–23]. There has been some debate over the number of different pulses of Neanderthal admixture, with recent evidence from ancient DNA suggesting it was a single admixture event as non-Africans migrated out of Africa [7,24]. It has also been suggested that the admixture event likely extended over thousands of years [7,25]. Interestingly, the total amount and the coverage of Neanderthal ancestry is reported to differ among non-African populations, with the reasons for this still an open question [26–30].
Evidence suggests that archaic ancestry was generally negatively selected against in the modern human genome, leaving large ‘deserts’ free of such ancestry [7,20,31–33]. Archaic ancestry has also been shown to be depleted on the X chromosome. These deserts are of great interest as potentially representing functionally important regions to modern humans (genes involved in brain development, immune function and reproduction have been suggested) or regions that create hybrid incompatibilities [16,20,34–37]. On the other hand, many studies have reported examples of adaptive introgression where archaic ancestry shows signals of positive selection, with genes involved in brain function, immunity, skin and hair pigmentation and the musculoskeletal system being suggested as candidates [38–45]. Work has also focused on understanding the functional consequences of archaic ancestry for modern humans, e.g., by using in vitro functional assays [46–48].
Here we compare the performance of three widely used methods, Sprime [13], Hmmix [18] and IBDmix [11] and one new method, cp-archaic, that is based on ChromoPainter and similar to previous local ancestry approaches [5,49,50]. Although these published methods are frequently used to explore introgressed archaic ancestry, there has been little attempt to benchmark the different approaches. Similarly, an in depth analysis of the properties of simulated and inferred segments is lacking. Here we present such an investigation, using both simulated data and real data from 1000 Genomes CHB and CEU populations. We also assess the extent to which different methods infer the same sites as archaic.
The new method introduced here, ‘cp-archaic’, utilises the ChromoPainter approach [51], and takes as input SNP data of individuals from each of an outgroup population assumed to not to have any archaic introgression, an archaic population(s), and a target population whose individuals potentially carry introgression from a source closely related to the archaic population(s). In analyses presented here, the outgroup population consists of 100 real or simulated African individuals, and the archaic population(s) consist of 1-4 real or simulated archaic individuals. Each target individual, and some of the individuals from the outgroup and archaic populations, are painted, as described in Lawson et al. 2012, against a set of individuals representing >=2 distinct ‘donor’ groups. These donor groups should be selected to distinguish between the outgroup and archaic populations; i.e., individuals from the outgroup and archaic populations should be most closely related genetically to separate donor groups. A natural choice is to use individuals from the archaic and outgroup populations themselves to define these donor groups, as we describe below.
There are two steps necessary when using cp-archaic. In the first step, cp-archaic uses the ChromoPainter approach to infer the total proportion of analysed genome for which individuals from the outgroup and archaic populations are painted against individuals from each of the donor groups. In the second step, for each haploid genome in the target population, cp-archaic uses the ChromoPainter approach to infer the probability that each SNP along the genome is painted against individuals from each of the same donor groups. As described below, cp-archaic combines these two steps to infer the probability that each SNP in a target haploid genome is inherited from the archaic population, and then uses these probabilities to infer introgressed archaic segments.
For the analyses presented here, in the first step one archaic individual, which we refer to as the ‘archaic surrogate’, and each of the N (e.g., N=100) outgroup individuals is painted against 2-4 donor groups. In the second step, each haploid genome of each target individual is painted against the same 2-4 donor groups. Here one of the donor groups consists of N-1 of the outgroup individuals, and the other 1-3 donor groups each consist of one of the 1-3 real or simulated archaic individuals that are not the archaic surrogate. We removed one outgroup individual from the donor set to ensure that both the archaic surrogate, outgroup and target individuals are painted by N-1 outgroup individuals (because an outgroup individual cannot be painted by themself). While we classify all outgroup individuals into a single donor population, we do not do the same for the archaics here, because the sampled archaic individuals are likely not as closely related to each other as the sampled outgroup individuals are, and hence perhaps should not be considered a single population. In simulations, we compare results when using different archaic individuals to define the archaic surrogate, and when using 1 versus 2 versus 3 of the other archaics to define the 1-3 archaic donor groups.
For analyses we report here, below we let O refer to the outgroup, A refer to the archaic surrogate and {D1,D2,D3} refer to the three donor archaic individuals that are not A. Let
We describe
where
We assume Pr(ancestry = A | painted by x) is the same at every SNP painted by x in every target haploid genome. In this case, for a target haploid genome, we define the probability that SNP s is inherited from the archaic population as:
where Pr(painted by x at SNP s) is inferred for each target haploid genome in the step two painting described above.
This protocol closely follows that described in van Dorp et al., 2015 [49]. However, in that paper they inferred analogues to
To call the final segments of archaic ancestry in a target haploid, the user specifies five parameters, three of which (c1,c2,c3) refer to basepair lengths and two of which (e1,e2) refer to probability thresholds. For brevity in this paragraph, let Pr(ancestry = A) = Pr(ancestry = A at SNP s). cp-archaic first identifies segments that are of length > c1 and contain only SNPs (minimum ten) with Pr(ancestry = A) ≥ e1. We provide possible choices of e1 below, but note that for any application e1 must be a value between the minimum and maximum of Pr(ancestry = A | painted by x) across x, since this defines the range of possible Pr(ancestry = A). With these segments ordered from left to right according to their position along the chromosome, let
We ran three separate demography simulations in msprime (Figures 1, S2, S3, S4, parameters in Text S1): one from Skov et al., 2018 simulating East Asian history with a unique bottleneck in the target population, one from Gower et al., 2021 simulating West Eurasian demography with a large recent increase in population size, and one with both Neanderthal and Denisovan introgression into a Papuan population, with parameters taken from the stdpopsim catalogue ‘Out-of-Africa with archaic admixture into Papuans’ but with non-Papuan populations removed [18,52–54]. For each demography, we simulated all 22 chromosomes using the standard plink recombination map in hg19, 200 target individuals and 100 ‘African’ outgroup/reference individuals (plus another 100 outgroup individuals only used as a phasing reference population). Simulated individuals had 30-36 million SNPs after filtering for biallelic sites only. For the two demographies with just Neanderthal admixture, we simulated two different archaic populations, each with two individuals sampled at 2330 generations ago (65,240ya assuming a 28-year generation time), which reflects the age of the Altai Neanderthal. One represents the ‘true’ introgressing population, and one a population that diverged from the introgressing population 130k years ago without migration. For each method, we tested the impact of using the introgressing or non-introgressing archaic as the archaic surrogate (see above). Additionally, we simulated 10 independent repeats of chromosome 1 of the Gower demography simulation to examine how repeatable archaic deserts and hotspots are, and if they correlate with recombination rate (Figure S5, S6).

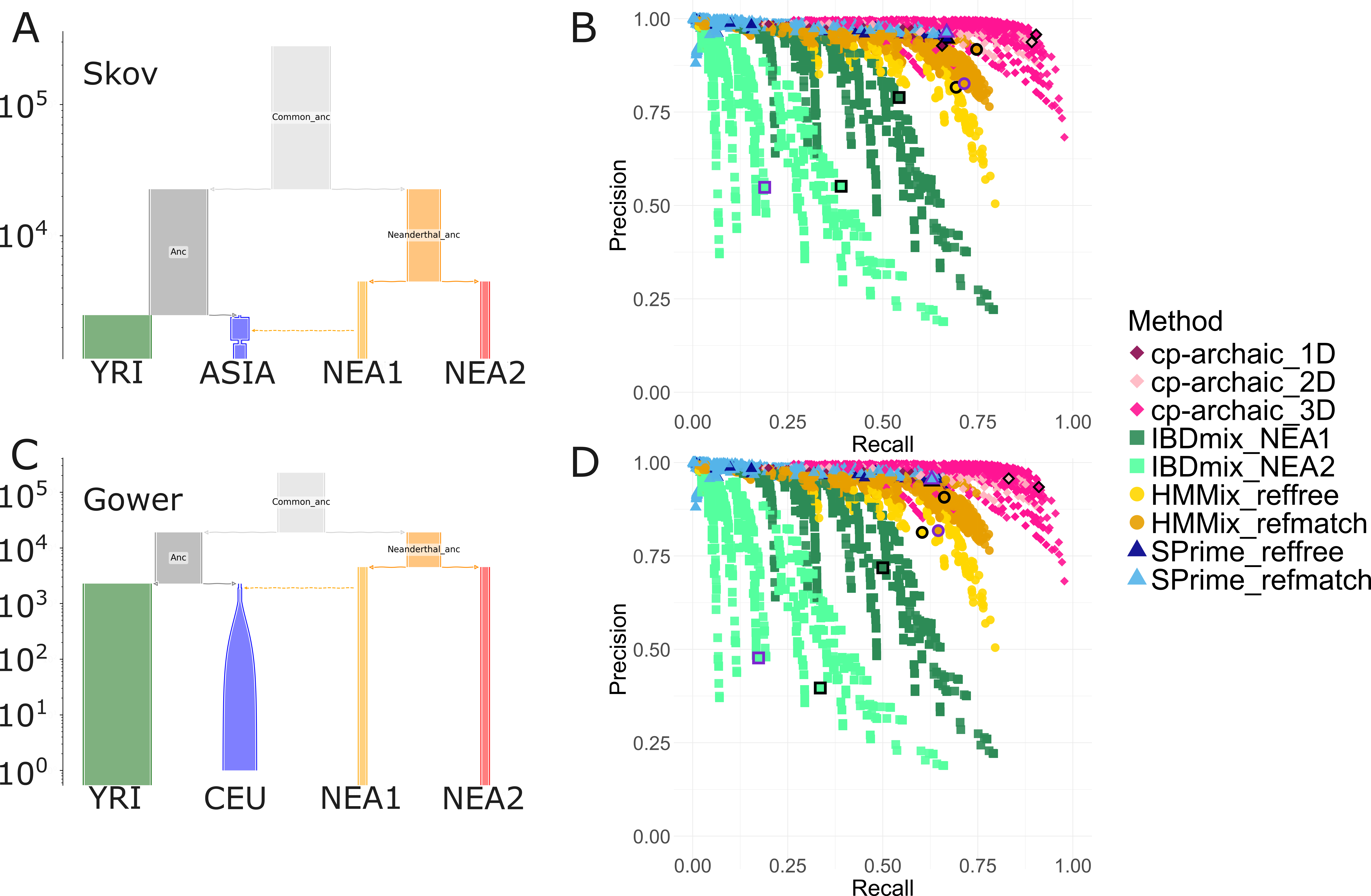
Figure 1. Performance of archaic introgression inference methods across different simulated demographies. Demographies used in (A) Skov et al., (2018) and (C) Gower et al (2021), simulated with msprime (parameters in Text S1). (B) and (D) recall and precision values for segments inferred for each demography simulation by the four different methods, cp-archaic, IBDmix, HMmix and SPrime. Each method was run under a variety of different parameters and filters, for example minimum length, score, match rate to archaics (see Methods). IBDmix was run using either one sample from the NEA1 (the introgressing Neanderthal) or NEA2 (the non-introgressing Neanderthal) population as a reference. cp-archaic was run with either one, two, or three sampled Neanderthals as ‘donors’ (1D, 2D, 3D) and always one (separate) Neanderthal as the archaic surrogate (see Methods). HMmix and SPrime were run either completely reference-free or filtering for match rate to the archaic genomes. Thin black borders indicate the best F1 score for the method, and thus the set of parameters used for future analyses of the simulation. Thick purple borders indicate ‘recommended’ or commonly used parameters for published methods. These two values are the same for cp-archaic.
We ran the four methods on the 200 simulated target individuals for each of the three demographies. For each method that requires an outgroup, we used 100 simulated outgroup individuals. HMmix was run using standard parameters in haploid mode (for phased data), following the tutorial provided with the paper (https://github.com/LauritsSkov/Introgression-detection) [18].
For SPrime, the results are given as a population-level summary of putatively archaic variants as a result of ‘tiling’ segments from different individuals together [13,14]. Therefore, to make SPrime comparable to other methods, we developed a pipeline to reconstruct individual-specific archaic segments. The approach takes the SPrime ‘.mscore’ file as input, as well as the VCF containing genotype calls for all individuals in the target population. The list of sites labeled as archaic by SPrime are mapped back onto the phased genotype data, keeping only the haplotypes that carry the SPrime-designated allele at a given site. For each individual and chromosome, we then defined candidate archaic segments using the following. We treated a sequence of archaic sites in a haplotype as belonging to a single continuous segment only if the distance between consecutive sites did not exceed 5kb; any gap larger than this threshold was interpreted as a potential break in the introgressed tract, initiating a new segment. This avoids over-fragmentation due to sparse markers or small gaps in coverage. We additionally explored merging neighbouring runs whose endpoints lay within a specified physical distance (10kb, 20kb, or 40kb), and assessed how this choice of merging threshold affected the inferred length distribution and accuracy of archaic tracts.
IBDmix was run with standard parameters, but setting the error rate to 0, using either the introgressing or the non-introgressing archaic as reference [11]. cp-archaic (as described above) was run with different numbers and sets of archaic donors (one, two, or three, including using the introgressing Neanderthal as a donor or not) and using one sample from either the introgressing population or the non-intogressing population as the archaic surrogate (defined above). For both true and inferred segments, we removed those that overlap the centromeric regions.
We then calculated precision and recall for each method under various parameter settings and filters, producing a distribution of values (Figure 1, S2). Recall and precision were calculated using base pair length overlap with the ‘true’ introgressing segments, without considering phase since IBDmix does not give haplotype information. To do this, we combine any segments that overlap on both haplotypes to create one longer segment, both for the ‘true’ segments and the inferred segments, before calculating total base pair overlap with the truth.
For all methods, we used different filters for the minimum length of archaic segments both in kilobases (0kb, 20kb, 30kb, 50kb, 100kb) and in centimorgans (0.01cM, 0.02cM, 0.05cM, 0.1cM). Additionally, we varied the minimum number of SNPs per segment (no filter, 10, 25, 50, 80, 150). For HMmix we varied the mean probability filter (0.5, 0.8, 0.9, 0.95, 0.99), for IBDmix we varied the LOD filter (3, 4, 8, 10, 15, 20, 25, 30, 40, 50, 100) and for cp-archaic we varied the value of e1 above (0.1, 0.5, 0.85). SPrime and HMmix infer segments without the use of a reference but allow matching to archaic references after running. Therefore, for these methods we also performed matching to one of: (i) either of the two sampled archaics from the introgressing population, (ii) either of the two sampled archaics from the non-introgressing population, or (iii) any of the four sampled archaics, in each case including filters based on the percentage of SNPs that match to the archaic reference in a segment (any match, 10%, 50%). For cp-archaic, we also varied the parameters c1,c2,c3 described above ((10kb,40kb,20kb), (10kb,20kb,20kb), (10kb,20kb,10kb), (0kb, 0kb, 0kb), (0kb, 0kb, 10kb), (0kb, 0kb, 20kb)). Considering all parameters combinations, for each simulation we calculated precision and recall for 4800 combinations for SPrime, 6000 combinations for HMmix, 4320 combinations for IBDmix and 3780 combinations for cp-archaic (Figure 1). The number of combinations reflects the number of different parameters that can be varied, and the number of options we chose. For example, for HMmix we have 6 genetic length filters, 5 length filters, 5 mean prob filters, 10 archaic filters and 4 site filters, leading to 6000 total combinations. For the Papuan demography simulation, we calculated overall precision and recall for any archaic segment, rather than specifically to Denisovan or Neanderthal. IBDmix and cp-archaic were run with either the simulated Neanderthal or Denisovan as reference/surrogate (Figure S2).
We defined a set of ‘best’ parameters as those that achieved the highest F1 score in the Skov demography (Data S1, see Data S2-3 for results from other demographies), where F1 = 2 * precision * recall / (precision + recall). We additionally defined a set of ‘recommended’ parameters from published papers for each method, although these may have some discrepancy as different papers can use different parameters. For the set of ‘best’ inferred segments, we additionally calculated the standard error for our estimates of precision and recall using both chromosome jackknifing and the formula from Busing et al., 1999 [55], and resampling with replacement 10 chromosomes for 100 repeats (Figure S7). We additionally examined the properties of the inferred segments, both at a haplotype level (Figure S8) and at a genotype level by merging segments across haplotypes (Figure S9). We then extracted the list of unique sites per individual (ignoring haplotype information) that each method infers to be introgressed, and compare the overlap of these (Figure S10). Finally, we calculated the match rate of all inferred segments from each method (without any length filters applied) to the ‘true’ segments, as percentage of the sequence length of inferred segments that overlaps with a true segment (Figure S11).
For cp-archaic, we tested the impact of changing the prior expectation of percentage archaic ancestry. We reran cp-archaic with a prior of 0.01, 0.02, 0.05 and 0.1 (Figure S12). Additionally, we investigated the impact of phasing errors on our results for the methods that require phased data. cp-archaic and HMmix both use phased input (although HMmix has an unphased mode too, not used here), and our SPrime post-processing pipeline utilises phased VCF data. We implemented two different phasing schemes, both using SHAPEIT5 [56]: (a) reference-free using ‘phase_common’ and (b) using 100 individuals from the target population and 100 individuals from the ‘outgroup’ population as a reference (with none of these individuals used in analysis). For (b) we ran the phasing in two steps, firstly running the ‘phase_common’ with –filter-maf 0.001. We then divided the genome into chunks using GLIMPSE2 [57], running ‘phase_rare’ on each chunk separately, before concatenating the chunks. We used both these schemes a) and b) to phase the 100 target individuals (which excludes any of those used as references for phasing), the 100 other outgroup individuals and 4 archaics that were used in downstream analyses to test the methods. We reran the three methods on the reference-free and reference phased simulated individuals, and compared the precision and recall to previous results using perfect phase for the same 100 target population individuals (Figure S13).
We compared the performance of our ‘cp-archaic’ approach to a previous approach using ChromoPainter, published in Jacobs et al., (2019) [5]. A difference between the two approaches is that cp-archaic uses a post-processing step that merges shorter segments into larger ones. Another difference is that the Jacobs et al., (2019) [5] approach uses 10 Expectation-Maximisation (E-M) steps to infer various ChromoPainter parameters when painting the target population individuals. These inferred parameters are the average rate of switching among donors, the average rate of allelic mismatching to a donor being painted by, and the average genome-wide probability of painting from each donor group. This E-M approach replaces the first step of cp-archaic described above that paints the outgroup and ancient surrogate individuals. However, by using steps of E-M, it takes ~10 times longer to paint each target individual over any given section of the genome. In practice only a subset of target individuals and genomic positions may need to be painted to infer the ChromoPainter parameters reliably via E-M, although Jacobs et al., 2019 [5] use the whole genome and all targets for their estimates (which should in theory give the best parameter estimates). Analogously, in practice only a subset of individuals may be required to run step 1 of cp-archaic, so that the time to run each approach may be similar in many applications. Here, to make the approaches comparable, we use all available data for step 1 of cp-archaic and for the E-M steps of the Jacobs approach, which results in cp-archaic being ~10-times faster in the simulations here.
We ran the Jacobs approach on chromosome 12-22 of the Skov simulated individuals (Figure S14). To run the Jacobs method, we first painted our target individuals using 100 outgroup individuals and the four archaic individuals as donors, for 10 EM steps (using parameters -i 10 -in -iM -ip) to estimate prior copying probabilities. Copying probabilities were estimated as follows: YRI_outgroup D 0.9755644, Nea1 D 0.008080701, Nea2 D 0.008205877, Nea3 D 0.004007203, Nea4 D 0.00414177, and Ne=7487.846 and Mu=0.007244771. We then ran the second step, painting the targets again using these inferred priors and parameters with the -b tag to create ‘copyprobsperlocus.out’ files. We converted this output into segments, by using an archaic threshold probability of 0.85 (as in Jacobs et al., 2019 [5]) and requiring a minimum of 10 consecutive SNPs exceeding this threshold. We compared the reliability and precision of this approach under several filtering conditions to cp-archaic.
Finally, we tested the performance of our new approach, cp-archaic, with a realistic mask file (using the one from Li et al, 2024 [12] which includes all archaic masks, described below) to understand if optimal parameters changed when >50% sites in the genome are masked. For the simulations of chromosome 2 and 15, we filtered out masked sites, reducing the number of available sites by 55%. We then tested the performance of cp-archaic using different e1 values (0.1, 0.5, 0.6, 0.8) and different minimum genetic length filters (none, 0.01cM, 0.02cM, 0.05cM). We found that with the mask file applied, the optimal filter conditions changed such that e1=0.6 and segments >0.02cM gave the highest F1 score (precision=0.97, recall=0.82, Data S4). Without masking, the optimal filter conditions were e1=0.1 and segments >0.01cM (precision=0.90, recall=0.96).
We compared the four methods’ inferred archaic segments in real data from CEU and CHB populations in the 1000 Genomes Project (Figure 2) [58]. For IBDmix and SPrime, we downloaded previously inferred archaic segments released by the method publishers, at [59] (from 2024, IBDmix, filtered following the ‘recommended’ parameter set) and [60] (from 2018, SPrime), while we ran the other two methods. For all methods, we applied the mask files used for running IBDmix (available at [61]) which include the union of the 2016 mask files for Altai, Vindija, Chagyrskaya and Denisovan, as well as masking the MHC region, CpG sites and within 5bp of indels [12]. The masks also included the 1000 Genomes strict mask, and a mask of segmental duplications. Additionally, we used the callset with the Altai Neanderthal as a reference, where possible. For example, for IBDmix we used the segments inferred with an Altai reference, and for HMmix and SPrime we filtered for higher match rate to Altai than Denisovan. This was important for our analysis in Figure 3, where the higher genome-wide coverage in CEU compared to CHB was previously reported for Neanderthal-specific SNPs only. For cp-archaic, we used Altai as the archaic surrogate, but included the other three archaics as donors.
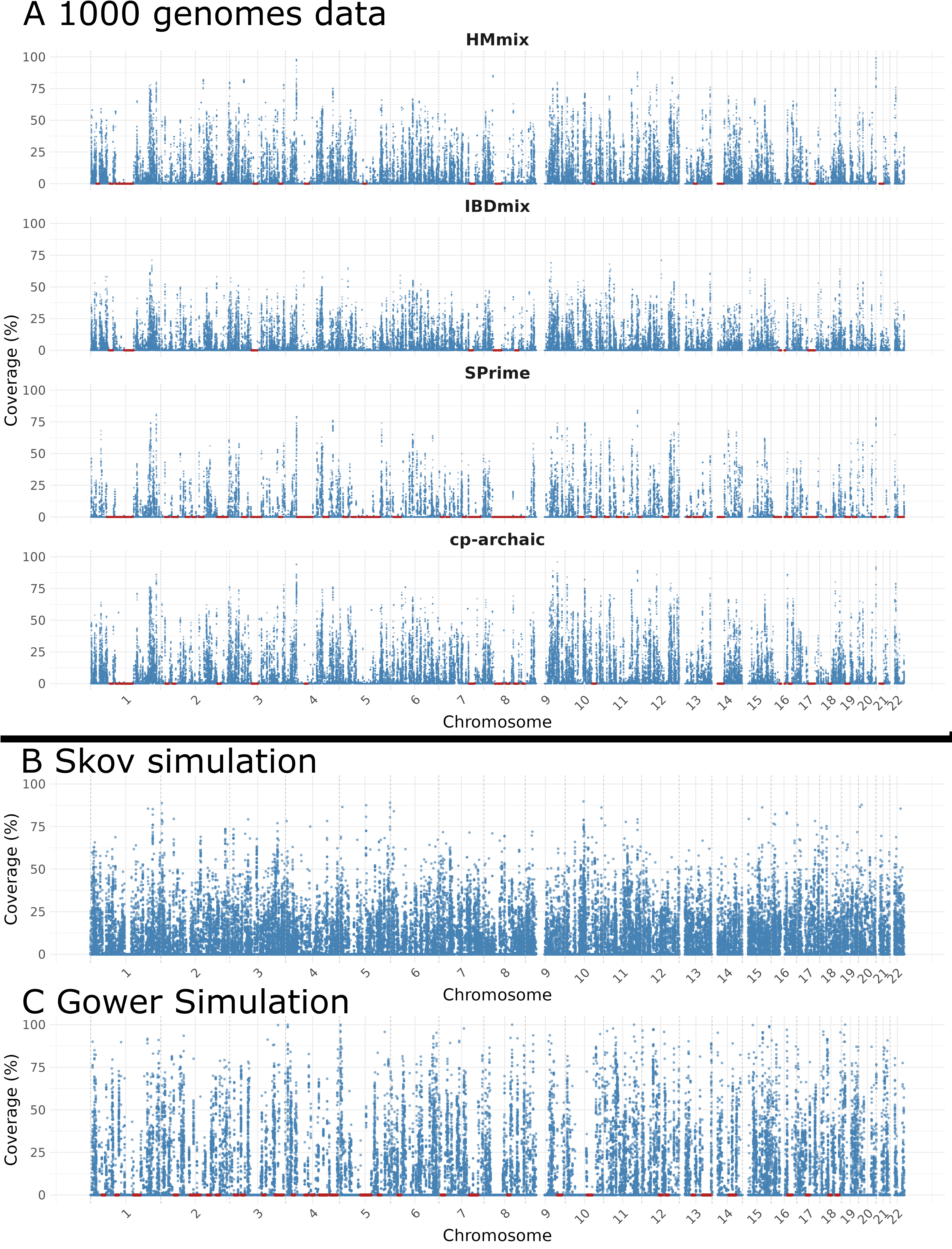
Figure 2. Manhattan plots of archaic coverage percentage (% of individuals with archaic inferred for at least one haplotype) in 103 CHB 1000 genomes individuals in 10kb windows for each of the four methods (rows). The final two rows show the true archaic coverage in the simulated individuals under the Skov and Gower simulation. Archaic deserts (region >=10Mb with <0.1% archaic ancestry) are highlighted in red, although these often overlap with centromeric regions. cp-archaic infers 22 deserts that do not overlap with the centromeres, IBDmix 9, HMmix 17 and SPrime 52. All deserts in the Skov simulation overlap with centromeric regions, but 41 from the Gower simulation do not.
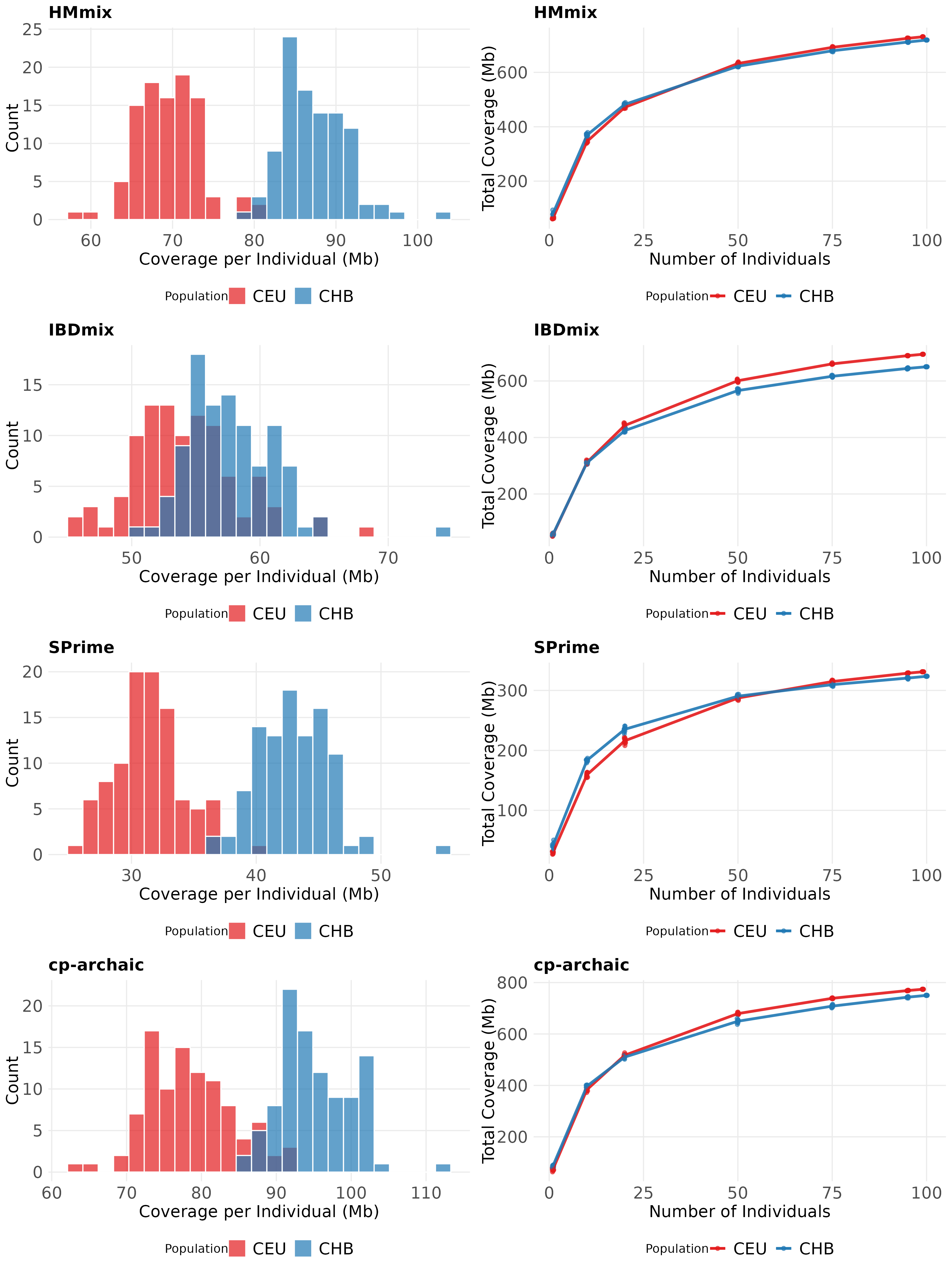
Figure 3. Individual and population-level archaic segment coverage distributions in CEU and CHB across inference methods. Coverage of inferred segments in CEU (red) and CHB (blue) for each of the methods cp-archaic (A), HMmix (B), IBDmix (C) and SPrime (D). The first column shows histograms of individual archaic coverage in each population, and the second column is total coverage when combining regions for which >=1 individual from the population is inferred to carry archaic DNA for different sub-sampled groups of individuals. To attempt to get Neanderthal specific segments, HMmix and SPrime segments were filtered for only segments that have a higher match rate to Altai Neanderthal than to Denisovan, and cp-archaic and IBDmix were run with the Altai Neanderthal as a reference/surrogate. However, these steps do not guarantee Denisovan segments do not remain to influence the coverage statistics.
SPrime from Browning et al., 2018 is run without using an archaic mask until the annotating archaic matching step. The annotated results available online had previously been run with just the 2016 Altai mask, so we additionally applied the more expansive IBDmix mask, annotating any sites excluded in the mask as ‘notcomp’. We then applied our additional script to match the SPrime-inferred archaic sites back to individual genotypes to get a final segment set (see description above). We merged individual segments <20kb apart, and filtered for segments with more matches to Altai Neanderthal than to Denisovan.
For HMmix, no segments were available online for 1000 Genomes data, so we ran it on CHB and CEU with 100 YRI individuals as an outgroup, following the example protocol included online [62] using the 1000 genomes strict mask. We ran HMmix decode using a vcf containing the Altai and Denisovan genomes, with the IBDmix masked sites excluded, and filtered to segments with a probability >0.9 and a higher match rate to the Altai Neanderthal than to the Denisovan (as explained above).
To run cp-archaic, we first phased the archaic individuals using SHAPEIT4 [63] with all populations in 1000 Genomes as a reference, in two steps, following the protocol in Speidel et al., 2025 [6]. First, all samples were phased at all SNPs except those with archaic-unique variants, using 1000 Genomes as a reference. Then this phasing was frozen as a scaffold to phase the remaining SNPs with archaic-unique variants. Next, we applied the IBDmix mask to the phased VCFs. We filtered the VCFs to exclude these sites before converting to ChromoPainter format, because cp-archaic relies on direct matching to the archaic genomes throughout so the mask must be applied first. We then ran cp-archaic on CEU and CHB using 100 YRI individuals as an outgroup (matching HMmix) and Vindija, Chagyrskaya and Denisovan as archaic donors, with the Altai Neanderthal as the archaic surrogate (defined above) [1,8–10]. cp-archaic was run on the phased data using (c1,c2,c3)=(10kb,40kb,20kb) and (e1,e2)=(0.6,0.01) and filtering for segments > 0.02cM (Figure S8).
We then extracted the list of unique sites per individual (ignoring haplotype information) that each method infers to be introgressed, and compare the overlap of these (Figure S15, S16). We compared only 1000 Genomes SNPs not included in the IBDmix mask files.
We first examined the distribution of true introgressed archaic segments in each of our demography simulations (Figure S3). We found a skewed distribution for all simulations, with the mean segment length (42-52kb) longer than the median segment length (12-16kb). Segments from the Gower simulation, where introgression occurs during the out of Africa bottleneck prior to a population size expansion in the admixed population, were longer than those from the Skov simulation, which simulates a bottleneck in the admixed population. Overall archaic ancestry percentage was less in the Gower simulation relative to the Skov simulation (1.8% vs 2%). In the Papuan simulation, the Denisovan segments were on average 10kb longer than the Neanderthal segments.
We also explored the distribution of simulated archaic ancestry across the genome (Figure 2, S4). We found differences between the demographies, with one striking observation being that the Gower simulation had 41 archaic ancestry deserts [20] (defined as regions >=10Mb with <0.1%, which in this case is 0% archaic ancestry, following Sankararaman et al., 2014 [20]) while the Skov simulation had 0. Taking all 200 simulated individuals together, the Skov simulation introgressed segments covered 578Mb of the genome, while the Gower simulation only covered 286Mb (Figure S4). However, the Gower simulation also had more windows with high (>90%) coverage out of all haploid genomes (Figure S3). We examined how random the position of these archaic deserts is by simulating 10 independent repeats of chromosome 1 of the Gower simulation (Figure S5). We found 33% of desert windows overlapped between >2 replicates, although the full deserts’ length and exact position varied. Desert windows with more overlaps among the 10 replicates tended to be in regions with lower mean recombination rate (Figure S6).
We then ran cp-archaic, IBDmix, HMmix and SPrime on each of the demography simulations to infer segments of archaic ancestry (Methods). For all methods, we combined different filter parameters, such as minimum length, segment score/probability and match rate to the archaic genomes, and calculated precision and recall by sequence length (see Methods; Figure 1, Figure S2). We found that the distribution of recall and precision values varied notably depending on the parameters picked, and between methods (Table 1). For the Skov simulation, the best F1 score (the harmonic mean of the recall and precision) ranged between 0.46 (IBDmix run with the non-introgressing Neanderthal, NEA2, as the reference) to 0.93 (cp-archaic run with three archaic donors). Overall, several different filtering schemes gave very similar F1 scores (see Data S1-3 for the top 10 parameter combinations). For example, for cp-archaic, while a segment merging scheme of (c1,c2,c3)=(10kb,40kb,20kb) (Methods, Figure S1) gave the best results, results without any merging of (c1,c2,c3)=(0kb,0kb,0kb) produced an F1 nearly as high (F1=0.93 vs 0.92, Data S1). We note we have tried to include ‘recommended’ parameter settings from previous publications. However, for HMmix different publications have used slightly different settings, and for HMmix and SPrime the match rate to archaic genomes is used as a secondary filtering step to identify high confidence segments. In this case, to compare to other methods, we filtered HMmix and SPrime for segments with any match to any of the four archaic genomes. Overall, cp-archaic with three archaic donors achieved the highest precision and recall. IBDmix’s performance varied drastically depending on if the introgressing archaic or the other archaic population was used as a reference. Interestingly, SPrime performed similarly with and without matching to an archaic reference. We calculated standard errors for our estimated recall and precision values using two separate methods that sub-sample the simulated data (Figure S7).
Table 1. Method performance summary under recommended and optimised parameters. Recall, precision and Mb per ind for each of the methods run on the Skov simulation with (A) recommended parameters (i.e., those used on 1000 genomes or in previous publications) and (B) the best performing parameters in our simulation.
We found that for the Gower demography simulation, both recall and precision were worse for all methods (Figure 1B vs 1D). For some methods, slightly different filtering conditions to those in Table 1 produced the best F1 score (Data S2). For example, for reference-free HMmix a score filter of 0.8 produced the best results for the Skov demography, and one of 0.9 was best for the Gower demography. For the simulation of Papuan demography, with both Neanderthal and Denisovan waves of introgression (Figure S2), we found that all methods performed similar to or better than with the other demographies. IBDmix performed particularly well when using the Denisovan genome as reference only, and the ‘best’ parameters for IBDmix were very different for this simulation (LOD >8, no length filter). Additionally, we found that the optimal parameters changed for cp-archaic when a mask file is applied (e=0.6, >0.02cM).
Under the ‘best’ filter conditions from the previous analysis (black outlined points, Figure 1) we found variation in the distribution of segments length, with cp-archaic inferring the longest mean and median segments, and HMmix the shortest (Figure S8). cp-archaic inferred the most archaic sequence per genome and IBDmix the least. We note that because IBDmix does not give haplotype information, the inferred segments may be longer than the other methods as a result of merging across haplotypes. Therefore, we also compared inferred segments at ‘genotype-level’ and found that HMmix still infers the shortest mean segments despite this (Figure S9). However, when comparing archaic sequence length per genome, at genotype level IBDmix and SPrime infer the same percentage.
We investigated the inferred segments from each method further by calculating the percentage overlap of all inferred segments from each method with a true segment in the same individual to analyse the properties of the most and least accurate segments (Figure S11). On the whole and as expected, longer inferred segments were more likely to be true positives, although this relationship was less strong in SPrime-inferred segments. Segments inferred by cp-archaic, HMmix and IBDmix had greater accuracy at higher recombination rates, and this relationship was stronger in shorter segments.
We tested the impact of phasing errors on cp-archaic and HMmix, which both require phased data to run, although HMmix has a genotype-only version not tested here (Figure S13). We also tested SPrime, which does not require phased data to run, but uses it in our post-processing matching step. With cp-archaic we found that imperfect phasing caused a small drop in precision (<0.01). For HMmix results did not change, except surprisingly the simulations with imperfect phasing had a small increase in recall. For SPrime, imperfect phasing had nearly no impact on the output. We also tried increasing the prior probability of archaic ancestry in cp-archaic and found a very small effect, with a higher prior percentage decreasing precision by <0.01 (Figure S13).
Finally, we compared the performance of cp-archaic with the other ChromoPainter-based method used in Jacobs et al., 2019 [5]. We found that both methods had a very similar performance as expected, although cp-archaic achieved slightly higher recall values (Figure S14). If the user has more target than donor individuals, cp-archaic will be ~10 times faster to run for the same segment of genome, as it does not require 10 EM steps to estimate copying probabilities. However, cp-archaic requires painting an additional set of outgroup and ancient surrogate individuals, which the Jacobs E-M approach does not. We note that the accuracy of both approaches is very similar, so that either could be run depending on the user’s needs (e.g., sample size of targets versus sample size of outgroup/ancients).
We analysed the site-by-site (SNP) overlap of archaic introgression calls for each simulated individual across the four methods (Figure S10, Table 2), under both our inferred best filtering conditions, and the recommended filters from published papers (Methods), for all chromosomes and 100 of the 200 simulated individuals from the Skov demography. We found that, using our filters, 24% of archaic sites called in any method were called in all methods, giving a recall of only 0.34 but a precision of 0.99, higher than any single method achieves. When using ‘recommended parameters’, this overlap was reduced to 20% with precision=0.99. We examined overlap among methods under a less stringent setting where we divided the genome into non-overlapping windows of 50kb or 1Mb, with each window classed as archaic if any inferred segment partially overlapped with it. Under this approach, the proportion of genome for which all four methods overlapped was still 24% for a window size of 50kb, but increased to 32% when using a window size of 1Mb. We also analysed population-level overlap between methods, comparing sites inferred to be archaic in any individual, and found that 28% of sites overlapped all four methods.
Table 2. Method-specific uniqueness of inferred archaic sites across simulations and 1000 Genomes data. Out of all sites inferred to be archaic by each method separately in the simulations and 1000 Genomes data, the percentage that are unique to that method (i.e., not inferred by any other method).
Next, we considered inferred segments in real data from the 1000 Genomes populations CEU and CHB populations using the four methods. Where it was available, for Sprime and IBDMix, we downloaded segments from published papers. In contrast, we ran HMmix ourselves following the online protocol. We attempted to ensure consistency between method runs by using the Altai Neanderthal as the reference/surrogate individual where possible. For example, for cp-archaic and IBDmix, we used the Altai Neanderthal as the archaic reference/surrogate, and for HMmix and SPrime we filtered such that inferred segments had a higher match rate to Altai than to the Denisovan in an attempt to remove Denisovan segments in CHB. However, while we used the Altai Neanderthal as the archaic surrogate in cp-archaic, we used the other three archaics as donors, potentially increasing power to detect archaic ancestry compared to the other methods. We applied the same archaic mask file to all methods.
We examined the distribution of segment lengths, coverage and proportion of archaic content inferred per individual in each of the methods and compared this to segments inferred from simulated data under the same filtering conditions (Figure S8). We found that all methods inferred a smaller percentage of the genome as archaic compared to the simulations, with the highest at 1.47% in CHB inferred with cp-archaic and lowest at 0.50% in CEU inferred with SPrime. Notably, SPrime inferred much less archaic ancestry in real data compared to the simulations (0.5% vs 1.4%). All methods also inferred on average longer segments in the real data, with for example HMmix inferring mean length to be more than double in real data (60kb vs 127kb). Coverage of the genome was different, with all methods inferring fewer segments that are found in a high percentage of the real individuals.
We analysed site by site overlap, overlap in windows of 1Mb, and population level overlap of inferred archaic ancestry by the four methods (Figure S15). Although we tried our best to ensure the same set of sites were analysed by each method by using the same mask file, we expect some potential discrepancy as a result of different quality control pipelines or callability masks. As in the simulations, we found a low percentage overlap at a SNP level between the four methods (18-21%). This overlap improved slightly if considering segments overlapping windows of 1Mb (19-24%) and at population level (29-31%). Overlap between methods was lower in CEU than CHB. IBDmix found the most unique sites in both populations, with SPrime inferring the least (Table 2).
We explored the coverage of archaic ancestry across the genome inferred by the four different methods (Figure 3). We attempted to filter out Denisovan segments in SPrime and HMmix by filtering for a greater match to Altai than Denisovan. IBDmix and cp-archaic were run using the Altai as reference/surrogate, but with no further filtering. Therefore, inferred segments from IBDmix and cp-archaic may potentially contain some Denisovan segments, though any such effect may be small given these two approaches do not infer more overlapping segments relative to other pairs of methods (Figure S15). We replicate previous findings that CHB individuals have more inferred archaic ancestry than CEU in all methods, although the difference in amount varied. For example, for IBDmix there was only a 4Mb difference in mean individual coverage, but in HMmix there was an 18Mb difference [11]. When combining archaic segments across individuals from each of the populations, we found total coverage (i.e., genomic regions covered by >=1 sampled individual) varied from 323-774Mb depending on population and method. While IBDmix and cp-archaic replicated previous findings that CHB have lower total coverage than CEU of archaic segments at a population level, SPrime and HMmix did not infer a clear pattern [26].
Archaic ancestry follows a peaked distribution, with many regions where >50% of individual genotypes in a population are inferred to have archaic ancestry (Figure 2, S16). A similar peaked pattern is also seen in the simulations, most strongly in the Gower simulation. Each method inferred a different number of archaic ancestry deserts (defined here as regions >=10Mb with <0.1% archaic ancestry [20]), with SPrime inferring the most (52 in CHB) and IBDmix the least (5 in CEU). All methods inferred more deserts in CHB than CEU.
Several different methods have been published to infer archaic ancestry segments in human genomes. Here, we explored the performance of a subset of these methods, choosing both reference-free and outgroup-free methods, as well as our new approach, cp-archaic (similar to Jacobs et al., 2019), which requires both. Overall, our results indicate that both the true underlying demographic scenario, method choice and the chosen parameters can alter conclusions about archaic ancestry in human genomes.
Different simulated demographies have a large impact on the distribution, amount and length of true segments. In the ‘Gower’ demography, archaic introgression occurs during the out of Africa bottleneck, followed closely by an increase in population size (Figure 1) [52]. This led to longer segments compared to the Skov simulation, where introgression occurred after the bottleneck [18], but also to fewer segments per individual and a more patchy coverage across the genome (Figure S3). Individuals simulated with the Gower demography had 41 archaic ancestry ‘deserts’ across the genome without any selection acting (Figure 2, Figure S4). These deserts occurred more often in regions of low recombination rate, where long segments inherited from a distant ancestor are more likely to be preserved. Additionally, both simulations have many regions with a high archaic ancestry proportion across haplotypes within a population (e.g., >50% in over 100Mb of the genome, Figure S3), as is also inferred in real data (Figure 2) [20]. Importantly, these results show how demography alone can create signals that could be interpreted as multiple waves of introgression or natural selection. The population size of modern humans during the introgression event may be particularly important. The underlying distribution of true segments can also change the recall and precision values of each method, and the optimal parameters (Figure 1, Data S1-3).
We also infer different properties in the simulated segments compared to the 1000 Genomes segments, for example they are shorter and have different coverage patterns (Figure S3 vs S8). This suggests that there are aspects of real archaic segments not well captured by our simulations which could significantly influence method performance. Previous papers have discussed the importance of simulation choice when understanding archaic introgression, showing that often patterns observed in real data do not reflect those in simulations well [26,64]. Other demographic factors not investigated here, such as generation time, mutation rate and population structure, may also be important considerations [29].
We demonstrate how the four different methods have very different recall and precision ranges within and across these simulated demographies, and these can vary significantly with parameter combination. Importantly, the ‘best’ result in all of these methods utilises an individual that is sampled from the simulated introgressing population ~10k years before the time of admixture, which we may not have access to in real data, rendering the highest possible values unrealistic. IBDmix is the only published method that does not require an ‘outgroup’. However, leveraging outgroup data, in addition to using multiple archaic reference genomes, might explain why other methods have higher recall and precision, at least in simulations with a genuinely unadmixed outgroup. In the real data, IBDmix infers a higher proportion of archaic sites in CEU that do not overlap with calls in any of the other three methods (26%, Table 2). While this may be attributable to introgressed segments in the African outgroup “masking” genuine segments in CEU not called by the other methods, caution is warranted given IBDmix also infers more unique sites in simulated data where no such “masking” occurs (Table 2).
Our new approach cp-archaic performs well, achieving an overall best F1 score of 0.93 in simulated data. It requires two or more archaic reference genomes and two or more outgroup individuals to run, making it specifically suited to inferring archaic ancestry in non-African genomes. In all the simulations we found that cp-archaic’s performance improved when using the introgressing Neanderthal (or reference Denisovan) as a donor (i.e., when painting other data against this archaic) rather than as the archaic surrogate (i.e., painting this archaic against other data). These results were only slightly impacted (<0.01) by imperfect (even completely reference-free) phasing of the outgroup, target and archaic genomes. Importantly, the method’s performance improved with an increased number of archaic donors, indicating increased power in the future as more high-coverage archaic genomes are published [65]. However, it requires an outgroup to run, making it less suited to infer the small amount of Neanderthal ancestry hypothesised to be present in African populations [11,66]. More generally, outside of archaic introgression, cp-archaic can be applied to infer local ancestry in admixed modern human groups as in similar techniques [49,50].
SPrime and HMmix are on the whole slightly less powerful than cp-archaic, even when forgoing their “reference-free” modes by matching inferred segments to the simulated archaics. This may be in part due to cp-archaic using archaic references when initially calling segments, compared to SPrime and HMmix initially calling segments without use of an archaic reference prior to post-hoc matching using the archaics. However, both can still achieve high precision and recall values. We find that filtering the reference-free methods on match rate to archaics improves their performance, but to a greater extent for HMmix than SPrime (HMmix best F1=0.82 with match filter, 0.75 without, SPrime best F1=0.79 with match filter, 0.78 without). We also find that for SPrime, segment accuracy is less influenced by the true basepair length of introgressed segments than in the other methods (Figure S11). We hypothesise that this is due to the fact that SPrime tiles archaic segments inferred from different individuals in a population, and here we have mapped these tiled segments back to individuals. As the tiling step borrows strength across individuals to infer segments, perhaps our mapping procedure gives SPrime increased power to infer shorter segments in individuals broken down by recombination.
On the whole, we find that longer segments tend to have a greater overlap with true segments, as expected given that longer segments tend to have more SNP data (Figure S11). Additionally, we find for all methods except SPrime, regions with lower recombination rate are more likely to be false positives, with the effect more pronounced in shorter segments. We hypothesise that some segments in low recombination regions result from incomplete lineage sorting (ILS) and are incorrectly called as introgressed. We find that for each of these methods, applying a genetic length filter improves performance. Applying such genetic length filters should become standard for inferring archaic segments in the future to mitigate these ILS effects. Previous work has shown that Neanderthal ancestry is more frequent in regions of higher recombination rate, potentially because it is more rapidly able to uncouple from deleterious alleles [67]. Our results show that this finding is likely to be robust to miscalling, as a greater number of false positives in low recombination regions would act to reduce this correlation.
A striking finding is the low overlap between the four methods in individual sites inferred to be archaic in both the simulated and real 1000 Genomes data (at 18-21% in 1000 Genomes, 20% in the simulations using similar filter parameters). Overlap like this would be expected if each of the four methods independently called an archaic region correctly ~67% of the time (0.67^4). Considering overlap among three of the four methods, the percentage overlap increases up to 52% in the simulation and <=55% in 1000 Genomes data. Even at a broader scale, in windows of 50kb and 1Mb, the overlap percentage does not increase significantly, suggesting the low overlap is not solely explained by differences in defining segment boundaries. Steinrücken et al., (2018) [4] previously suggested using the overlap between multiple methods to identify highly confident calls. Our findings support this conservative approach if highly confident segments are required, as a precision of 0.99 can be achieved at the intersection of all four methods. The substantial disagreement between methods suggests that individual archaic ancestry calls should be interpreted with caution. Additionally, we found that concordance between methods improves when comparing at the population-level rather than at the individual level. This likely reflects the lower detection power at an individual level, and suggests an approach like SPrime which borrows power across individuals in a population could yield better results.
The different overlap percentages in real data compared to the simulations (Table 2), even when using the same parameter set, suggests that there are aspects of real archaic segments not captured in the simulations which are influencing method performance. Some of this discrepancy may be explained by different analysis pipelines for each method, for example using different recombination rate maps and quality control filters. Additionally, for IBDmix, SPrime and HMmix we used calls made using the Altai Neanderthal, while cp-archaic utilised all four available high-coverage archaic genomes, giving it increased power. We note that at the moment, Vindija is the closest reference to the introgressing Neanderthal [10] and therefore leads to methods calling more introgressed sequence and potentially resulting in increased accuracy. However, incorporating multiple reference genomes, as implemented in cp-archaic, is expected to improve overall detection power by capturing a broader spectrum of archaic diversity.
Results from all four methods confirm previous findings that CHB individuals have more Neanderthal ancestry than CEU, although the magnitude of this difference depends on the method used [11,68]. Importantly, we demonstrate that conclusions about the total population-wide coverage of archaic ancestry are method dependent (Figure 3). For example, despite both HMmix and SPrime being filtered for segments with a higher match rate to Neanderthal than Denisovan, they show very little difference between the level of genome-wide archaic coverage among CHB versus CEU individuals. Further research is needed to understand the underlying causes of these methodological disagreements.
Archaic introgression has emerged as a rapidly expanding field of research, with increasingly innovative approaches to address the links between introgression, evolution and health. Powerful new methods are being published, including those specialised to identify segments introgressed from different archaic populations or specifically in admixed individuals descending from populations with different amounts of introgression [19,69,70]. The possibility of inferring archaic ancestry in ancient individuals is increasingly being explored, with a recent study finding imputed ancient genomes perform well for inferring archaic ancestry [7,71]. At the same time, studies inferring archaic ancestry in increasingly larger datasets of modern individuals are being published [27,31,72]. However, our findings highlight critical considerations for the field: demographic assumptions underlying simulations can profoundly influence conclusions about archaic introgression patterns, including insights into selection, method choice and parameter optimisation are crucial for reliable inference, and the substantial disagreement between methods calls for caution when interpreting individual archaic segments. As the field continues to expand and tackle increasingly complex datasets, careful validation of methodological approaches will be essential for ensuring robust findings.
The following supplementary materials are available on the website of this paper at HPGG2606020007SupplementaryMaterials.zip.
Figure S1. Cartoon depicting the segment merging scheme used in cp-archaic (Methods).
Figure S2. Performance of archaic segment detection methods under a Papuan-like simulated demography.
Figure S3. Summary of ‘true’ archaic segments from the three demographic simulations in Figures 1 and S1.
Figure S4. Individual and population-level archaic segment coverage distributions of simulated segments under different demographies. Figure S5. Manhattan plots from 10 independent simulations of chromosome 1 of the Gower demography.
Figure S6. Relationship between local recombination rate and frequency of overlap with simulated archaic desert regions.
Figure S7. Uncertainty in recall and precision estimates.
Figure S8. Summary of inferred archaic segments, using the four methods in simulated (top) and real (bottom) data.
Figure S9. Summary of inferred archaic segments, using the four methods and the ‘published’ parameters, where segments are considered at a haplotype level (lighter colours, right) and a genotype-level (merged across haplotypes, darker colors, left).
Figure S10. Overlap of archaic SNP calls across methods under optimized and published filtering schemes at SNP, window, and population levels in the Skov simulation. Figure S11. Accuracy of inferred archaic segments across recombination rate and length bins for different methods.
Figure S12. The performance (recall and precision) of cp-archaic when varying the prior probability of archaic ancestry on the Skov simulation (2% was used for all analyses on simulated individuals).
Figure S13. Impact of phasing strategy on precision and recall for phased-based archaic inference methods in the Skov simulation.
Figure S14. Comparing the performance (recall and precision) of cp-archaic with the ChromoPainter-based method used in Jacobs et al., (2019) for chromosomes 12-22 under the Skov demography (see Methods for a description of the approaches).
Figure S15. Overlap of archaic SNP calls across methods in 1000 Genomes CEU and CHB populations at SNP, window (1Mb), and population levels.
Figure S16. Manhattan plots of archaic coverage percentage in 98 CEU 1000 genomes individuals in 10kb windows for each of the four methods (rows).
Data S1. Top 10 best F1 scores for each of the four methods under different filtering schemes and the Skov demographic simulation.
Data S2. Top 10 best F1 scores for each of the four methods under different filtering schemes and the Gower demographic simulation.
Data S3. Top 10 best F1 scores for each of the four methods under different filtering schemes and the Denisovan demographic simulation.
Data S4. Top 5 best F1 scores for cp-archaic applied to chromosome 2 and 15 of masked data, under different filtering schemes and the Skov demographic simulation.
Text S1. Msprime code used to simulate each demography.
Not applicable.
Not applicable.
Cp-archaic is available online: https://github.com/nancybird/cp-archaic
Other code and data used in this manuscript is available here: https://github.com/nancybird/reliability-of-inferred-archaic-segments
This study was supported by the funding of Wellcome Trust (224575/Z/21/Z).
The authors have declared that no competing interests exist.
Conceptualization: NB and GH. Formal analysis: NB and EW. Methodology and Software: NB and GH. Writing- Original Draft: NB. Writing- Review and Editing: all.
We thank Dr. Leo Speidel for his suggestions on this work, as well as Prof. Joshua Akey and three anonymous reviewers for their helpful comments on the manuscript. Additionally, we thank colleagues for helpful discussions at a poster presentation at the SMBE Satellite Meeting (Ancient DNA Beyond Allele Frequencies, Dublin 2024).
| 1. | Meyer M, Kircher M, Gansauge MT, Li H, Racimo F, Mallick S, et al. A high-coverage genome sequence from an archaic Denisovan individual. Science. 2012;338(6104):222-226. [Google Scholar] [CrossRef] |
| 2. | Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, et al. A draft sequence of the Neandertal genome. Science. 2010;328(5979):710-722. [Google Scholar] [CrossRef] |
| 3. | Sankararaman S, Patterson N, Li H, Pääbo S, Reich D. The Date of Interbreeding between Neandertals and Modern Humans. PLoS Genet. 2012;8(10):e1002947. [Google Scholar] [CrossRef] |
| 4. | Steinrücken M, Spence JP, Kamm JA, Wieczorek E, Song YS. Model-based detection and analysis of introgressed Neanderthal ancestry in modern humans. Mol Ecol. 2018;27(19):3879-3902. [Google Scholar] [CrossRef] |
| 5. | Jacobs GS, Hudjashov G, Saag L, Kusuma P, Darusallam CC, Lawson DJ, et al. Multiple Deeply Divergent Denisovan Ancestries in Papuans. Cell. 2019;177(4):1010-1021.e32. [Google Scholar] [CrossRef] |
| 6. | Speidel L, Silva M, Booth T, Raffield L, Anastasiadou K, Barrie W, et al. High-resolution genomic history of early medieval Europe. Nature. 2025;637(8045):118-126. [Google Scholar] [CrossRef] |
| 7. | Iasi LNM, Boskovic M, Borić D, Pavlović M, Marković J, Blagojević T, et al. Neanderthal ancestry through time: Insights from genomes of ancient and present-day humans. Science. 2024;386(6727):eadq3010. [Google Scholar] [CrossRef] |
| 8. | Mafessoni F, Grote S, de Filippo C, Slon V, Kolobova KA, Viola B, et al. A high-coverage Neandertal genome from Chagyrskaya Cave. Proc Natl Acad Sci USA. 2020;117(26):15132-15136. [Google Scholar] [CrossRef] |
| 9. | Prüfer K, Racimo F, Patterson N, Jay F, Sankararaman S, Sawyer S, et al. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature. 2014;505(7481):43-49. [Google Scholar] [CrossRef] |
| 10. | Prüfer K, de Filippo C, Grote S, Mafessoni F, Korlević P, Hajdinjak M, et al. A high-coverage Neandertal genome from Vindija Cave in Croatia. Science. 2017;358(6363):655-658. [Google Scholar] [CrossRef] |
| 11. | Chen L, Wolf AB, Fu W, Li L, Akey JM. Identifying and Interpreting Apparent Neanderthal Ancestry in African Individuals. Cell. 2020;180(4):677-687.e16. [Google Scholar] [CrossRef] |
| 12. | Li L, Comi TJ, Bierman RF, Akey JM. Recurrent gene flow between Neanderthals and modern humans over the past 200,000 years. Science. 2024;385(6708):eadi1768. [Google Scholar] [CrossRef] |
| 13. | Browning SR, Browning BL, Zhou Y, Tucci S, Akey JM. Analysis of Human Sequence Data Reveals Two Pulses of Archaic Denisovan Admixture. Cell. 2018;173(1):53-61.e9. [Google Scholar] [CrossRef] |
| 14. | Zhou Y, Browning SR. Protocol for detecting introgressed archaic variants with SPrime. STAR Protoc. 2021;2(2):100550. [Google Scholar] [CrossRef] |
| 15. | Plagnol V, Wall JD. Possible ancestral structure in human populations. PLoS Genet. 2006;2(7):e105. [Google Scholar] [CrossRef] |
| 16. | Vernot B, Tucci S, Kelso J, Schraiber JG, Wolf AB, Gittelman RM, et al. Excavating Neandertal and Denisovan DNA from the genomes of Melanesian individuals. Science. 2016;352(6282):235-239. [Google Scholar] [CrossRef] |
| 17. | Huang X, Kruisz P, Kuhlwilm M. Sstar: A python package for detecting archaic introgression from population genetic data with S. Mol Biol Evol. 2022;39(10):msac212. [Google Scholar] [CrossRef] |
| 18. | Skov L, Hui R, Shchur V, Hobolth A, Scally A, Schierup MH, et al. Detecting archaic introgression using an unadmixed outgroup. PLoS Genet. 2018;14(9):e1007641. [Google Scholar] [CrossRef] |
| 19. | Planche L, Ilina AV, Shchur VL. Highly accurate method for detecting archaic segments in the modern genomes. Lobachevskii J Math. 2024;45(6):2910-2917. [Google Scholar] [CrossRef] |
| 20. | Sankararaman S, Mallick S, Dannemann M, Prüfer K, Kelso J, Pääbo S, et al. The genomic landscape of Neanderthal ancestry in present-day humans. Nature. 2014;507(7492):354-357. [Google Scholar] [CrossRef] |
| 21. | Mondal M, Bertranpetit J, Lao O. Approximate Bayesian computation with deep learning supports a third archaic introgression in Asia and Oceania. Nat Commun. 2019;10(1):246. [Google Scholar] [CrossRef] |
| 22. | Ongaro L, Huerta-Sanchez E. A history of multiple Denisovan introgression events in modern humans. Nat Genet. 2024;56(12):2612-2622. [Google Scholar] [CrossRef] |
| 23. | Larena M, McKenna J, Sanchez-Quinto F, Bernhardsson C, Ebeo C, Reyes R, et al. Philippine Ayta possess the highest level of Denisovan ancestry in the world. Curr Biol. 2021;31(19):4219-4230.e6. [Google Scholar] [CrossRef] |
| 24. | Sümer AP, Rougier H, Villalba-Mouco V, Huang Y, Coll Macià M, Essel E, et al. Earliest modern human genomes constrain timing of Neanderthal admixture. Nature. 2025;638(8051):711-717. [Google Scholar] [CrossRef] |
| 25. | Iasi LNM, Ringbauer H, Peter BM. An extended admixture pulse model reveals the limitations to human-Neandertal introgression dating. Mol Biol Evol. 2021;38(11):5156-5174. [Google Scholar] [CrossRef] |
| 26. | Witt KE, Villanea F, Loughran E, Zhang X, Huerta-Sanchez E. Apportioning archaic variants among modern populations. Philos Trans R Soc Lond B Biol Sci. 2022;377(1852):20200410. [Google Scholar] [CrossRef] |
| 27. | Kerdoncuff E, Skov L, Coll Macià M, Guan Y, Marnetto D, Moorjani P, et al. 50,000 years of evolutionary history of India: Impact on health and disease variation. Cell. 2025;188(12):3389-3404.e6. [Google Scholar] [CrossRef] |
| 28. | Kim BY, Lohmueller KE. Selection and reduced population size cannot explain higher amounts of Neandertal ancestry in East Asian than in European human populations. Am J Hum Genet. 2015;96(3):454-461. [Google Scholar] [CrossRef] |
| 29. | Coll Macià M, Skov L, Peter BM, Schierup MH. Different historical generation intervals in human populations inferred from Neanderthal fragment lengths and mutation signatures. Nat Commun. 2021;12(1):1-11. [Google Scholar] [CrossRef] |
| 30. | Villanea FA, Schraiber JG. Multiple episodes of interbreeding between Neanderthal and modern humans. Nat Ecol Evol. 2019;3(1):39-44. [Google Scholar] [CrossRef] |
| 31. | Skov L, Macià MC, Sveinbjörnsson G, Mafessoni F, Lucotte EA, Einarsdóttir MS, et al. The nature of Neanderthal introgression revealed by 27,566 Icelandic genomes. Nature. 2020;582(7810):78-83. [Google Scholar] [CrossRef] |
| 32. | Yuan K, Ni X, Liu C, Pan Y, Deng L, Zhang R, et al. Refining models of archaic admixture in Eurasia with ArchaicSeeker 2.0. Nat Commun. 2021;12(1):6232. [Google Scholar] [CrossRef] |
| 33. | Petr M, Pääbo S, Kelso J, Vernot B. Limits of long-term selection against Neandertal introgression. Proc Natl Acad Sci USA. 2019;116(5):1639-1644. [Google Scholar] [CrossRef] |
| 34. | Buisan R, Moriano J, Andirkó A, Boeckx C. A brain region-specific expression profile for genes within large introgression deserts and under positive selection in Homo sapiens. Front Cell Dev Biol. 2022;10:824740. [Google Scholar] [CrossRef] |
| 35. | Ragsdale AP. Archaic introgression and the distribution of shared variation under stabilizing selection. PLoS Genet. 2025;21(2):e1011623. [Google Scholar] [CrossRef] |
| 36. | Harris K, Nielsen R. The genetic cost of Neanderthal introgression. Genetics. 2016;203(2):881-891. [Google Scholar] [CrossRef] |
| 37. | Telis N, Aguilar R, Harris K. Selection against archaic hominin genetic variation in regulatory regions. Nat Ecol Evol. 2020;4(11):1558-1566. [Google Scholar] [CrossRef] |
| 38. | Zhang X, Witt KE, Villanea FA, Loughran E, Huerta-Sanchez E. The history and evolution of the Denisovan-EPAS1 haplotype in Tibetans. Proc Natl Acad Sci USA. 2021;118(22):e2020803118. [Google Scholar] [CrossRef] |
| 39. | Huerta-Sánchez E, Jin X, Asan Bianba Z, Peter BM, Vinckenbosch N, et al. Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature. 2014;512(7513):194-197. [Google Scholar] [CrossRef] |
| 40. | Reilly PF, Tjahjadi A, Miller SL, Akey JM, Tucci S. The contribution of Neanderthal introgression to modern human traits. Curr Biol. 2022;32(18):R970-R983. [Google Scholar] [CrossRef] |
| 41. | Racimo F, Sankararaman S, Nielsen R, Huerta-Sánchez E. Evidence for archaic adaptive introgression in humans. Nat Rev Genet. 2015;16(6):359-371. [Google Scholar] [CrossRef] |
| 42. | Gittelman RM, Schraiber JG, Vernot B, Mikacenic C, Wurfel MM, Akey JM. Archaic hominin admixture facilitated adaptation to out-of-Africa environments. Curr Biol. 2016;26(24):3375-3382. [Google Scholar] [CrossRef] |
| 43. | Tagore D, Akey JM. Archaic hominin admixture and its consequences for modern humans. Curr Opin Genet Dev. 2025;90:102280. [Google Scholar] [CrossRef] |
| 44. | Villanea FA, Loughran E, Zhang X, Witt KE, Fielder J, Huerta-Sanchez E. The MUC19 gene: An evolutionary history of recurrent introgression and natural selection. Science. 2025;389(6748):eado5329. [Google Scholar] [CrossRef] |
| 45. | Wei X, Robles CR, Pazokitoroudi A, Ganna A, Gusev A, Durvasula A, et al. The lingering effects of Neanderthal introgression on human complex traits. Elife. 2023;12:e80757. [Google Scholar] [CrossRef] |
| 46. | Findley AS, Monzón-Sandoval J, Campanale A, Sattar A, Hodges E, Fuentes DR, et al. A signature of Neanderthal introgression on molecular mechanisms of environmental responses. PLoS Genet. 2021;17(8):e1009493. [Google Scholar] [CrossRef] |
| 47. | Jagoda E, Marnetto D, Mégevand A, Grenier JC, Roux M, Larbi A, et al. Regulatory dissection of the severe COVID-19 risk locus introgressed by Neanderthals. Elife. 2023;12:e80756. [Google Scholar] [CrossRef] |
| 48. | Zeberg H, Jakobsson M, Pääbo S. The genetic changes that shaped Neandertals, Denisovans, and modern humans. Cell. 2024;187(5):1047-1058. [Google Scholar] [CrossRef] |
| 49. | van Dorp L, Balding D, Myers S, Pagani L, Tyler-Smith C, Bekele E, et al. Evidence for a Common Origin of Blacksmiths and Cultivators in the Ethiopian Ari within the Last 4500 Years: Lessons for Clustering-Based Inference. PLoS Genet. 2015;11(8):e1005393. [Google Scholar] [CrossRef] |
| 50. | Molinaro L, Montinaro F, Gozzoli A, Pagani L, Capodiferro MR, Colangelo A, et al. A Chromosome-Painting-Based Pipeline to Infer Local Ancestry under Limited Source Availability. Genome Biol Evol. 2021;13(4):evab025. [Google Scholar] [CrossRef] |
| 51. | Lawson DJ, Hellenthal G, Myers S, Falush D. Inference of population structure using dense haplotype data. PLoS Genet. 2012;8(1):e1002453. [Google Scholar] [CrossRef] |
| 52. | Gower G, Picazo PI, Fumagalli M, Racimo F. Detecting adaptive introgression in human evolution using convolutional neural networks. Elife. 2021;10:e64669. [Google Scholar] [CrossRef] |
| 53. | Lauterbur ME, Cavassim MIA, Gladstein AL, Gower G, Pope NS, Tsambos G, et al. Expanding the stdpopsim species catalog, and lessons learned for realistic genome simulations. Elife. 2023;12:e84874. [Google Scholar] [CrossRef] |
| 54. | Baumdicker F, Bisschop G, Goldstein D, Gower G, Ragsdale AP, Tsambos G, et al. Efficient ancestry and mutation simulation with msprime 1.0. Genetics. 2022;220(3):iyab229. [Google Scholar] [CrossRef] |
| 55. | Busing FMTA, Meijer E, van der Leeden R. Delete-m Jackknife for Unequal m. Stat Comput. 1999;9(1):3-8. [Google Scholar] [CrossRef] |
| 56. | Hofmeister RJ, Ribeiro DM, Rubinacci S, Delaneau O. Accurate rare variant phasing of whole-genome and whole-exome sequencing data in the UK Biobank. Nat Genet. 2023;55(7):1243-1249. [Google Scholar] [CrossRef] |
| 57. | Rubinacci S, Hofmeister RJ, Sousa da Mota B, Delaneau O. Imputation of low-coverage sequencing data from 150,119 UK Biobank genomes. Nat Genet. 2023;55(7):1088-1090. [Google Scholar] [CrossRef] |
| 58. | 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015;526(7571):68-74. [Google Scholar] [CrossRef] |
| 59. | Princeton University / IBDmix [Internet]. GitHub. [cited 2023 Jul 19]. Available from: https://github.com/PrincetonUniversity/IBDmix. |
| 60. | Browning S. Sprime results for 1000 Genomes non-African populations and SGDP Papuans [Internet]. Mendeley Data; 2018 [cited 2023 Aug 21]. Available from: https://data.mendeley.com/datasets/y7hyt83vxr/1. [CrossRef] |
| 61. | Index of/AKEY/IBDmix_EXCLUDE_masks_hg19_Li_et_al_Science_2024 [Internet]. Princeton University Tigress. [cited 2025 Nov 19]. Available from: https://tigress-web.princeton.edu/AKEY/IBDmix_EXCLUDE_masks_hg19_Li_et_al_Science_2024/. |
| 62. | LauritsSkov / Introgression-detection [Internet]. GitHub. [cited 2023 Jul 17]. Available from: https://github.com/LauritsSkov/Introgression-detection. |
| 63. | Delaneau O, Zagury JF, Robinson MR, Marchini JL, Dermitzakis ET. Accurate, scalable and integrative haplotype estimation. Nat Commun. 2019;10(1):5436. [Google Scholar] [CrossRef] |
| 64. | Tournebize R, Chikhi L. Ignoring population structure in hominin evolutionary models can lead to the inference of spurious admixture events. Nat Ecol Evol. 2025;9(2):225-236. [Google Scholar] [CrossRef] |
| 65. | Peyrégne S, Slon V, Mafessoni F, de Filippo C, Hajdinjak M, Nagel S, et al. A high-coverage genome from a 200,000-year-old Denisovan. bioRxiv. 2025;2025.10.20.683404. [Google Scholar] [CrossRef] |
| 66. | Harris DN, Platt A, Hansen MEB, Fan S, Trask M, Koenig Z, et al. Diverse African genomes reveal selection on ancient modern human introgressions in Neanderthals. Curr Biol. 2023;33(22):4905-4916.e5. [Google Scholar] [CrossRef] |
| 67. | Schumer M, Xu C, Powell DL, Durvasula A, Skov L, Holland C, et al. Natural selection interacts with recombination to shape the evolution of hybrid genomes. Science. 2018;360(6389):656-660. [Google Scholar] [CrossRef] |
| 68. | Juric I, Aeschbacher S, Coop G. The strength of selection against Neanderthal introgression. PLoS Genet. 2016;12(11):e1006340. [Google Scholar] [CrossRef] |
| 69. | Planche L, Ilina AV, Shchur V, Zaporozhchenko K, Pankova A, Kolesnikov A, et al. An archaic reference-free method to jointly infer Neanderthal and Denisovan introgressed segments in modern human genomes. bioRxiv. 2025;2025.03.17.643330. [Google Scholar] [CrossRef] |
| 70. | Macià MC, Skov L, Damgaard Bæk ZE, Hobolth A. Enhancement of hidden Markov model analyses for improved inference of archaic introgression in modern humans. bioRxiv. 2025;2025.04.22.649993. [Google Scholar] [CrossRef] |
| 71. | Capodiferro MR, Raveane A, Colombo E, Ferretti L, Montinaro F, Migliore NR, et al. Archaic ancestry inference in imputed ancient human genomes. bioRxiv. 2025;2025.07.23.664192. [Google Scholar] [CrossRef] |
| 72. | Liu X, Koyama S, Tomizuka K, Ogawa K, Sawada Y, Ishikawa T, et al. Decoding triancestral origins, archaic introgression, and natural selection in the Japanese population by whole-genome sequencing. Sci Adv. 2024;10(17):eadi8419. [Google Scholar] [CrossRef] |

![]()
Copyright © 2026 Pivot Science Publications Corp. - unless otherwise stated | Terms and Conditions | Privacy Policy




