
Cancer Heterogeneity and Plasticity ISSN 2818-7792
Cancer Heterogeneity and Plasticity 2025;2(1):0003 | https://doi.org/10.47248/chp2502010003
Review Open Access
DIAPH3 in Cancer: Role and Mechanism
Jiangling Xiong
1,2,†
 ,
Lanxin Hu
1,†
,
Jinwei Zhu
1
,
Lianlian Yan
1
,
Yuqing Feng
1
,
Cheng Zou
1
,
Yang Mei
1
,
Dinglan Wu
3
,
Dingxiao Zhang
1,2
,
Lanxin Hu
1,†
,
Jinwei Zhu
1
,
Lianlian Yan
1
,
Yuqing Feng
1
,
Cheng Zou
1
,
Yang Mei
1
,
Dinglan Wu
3
,
Dingxiao Zhang
1,2
Correspondence: Yang Mei; Dinglan Wu; Dingxiao Zhang
Academic Editor(s): Anna Dubrovska
Received: Dec 6, 2024 | Accepted: Feb 15, 2025 | Published: Feb 25, 2025
© 2025 by the author(s). This is an Open Access article distributed under the terms of the Creative Commons License Attribution 4.0 International (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly credited.
Cite this article: Xiong J, Hu L, Zhu J, Yan L, Feng Y, Zou C, Mei Y, Wu D, Zhang D. DIAPH3 in Cancer: Role and Mechanism. Cancer Heterog Plast. 2025;2(1):0003. https://doi.org/10.47248/chp2502010003
Diaphanous-related formin 3 (DIAPH3) is a pivotal member of the formin family and serves as a crucial regulator of actin filament assembly. As such, DIAPH3 plays an integral role in a variety of cellular processes including cytokinesis, cell migration, and intracellular transport. Given its fundamental importance in maintaining cytoskeletal dynamics, DIAPH3 is functionally associated with numerous physiological and pathological conditions, particularly cancer. In this review, we explore the structural and functional characteristics of DIAPH3 and investigate its mutational and transcriptional landscape in human cancers. By focusing on DIAPH3’s role in controlling metastasis and tumor microenvironment, we aim to provide new insights into how DIAPH3 contributes to tumor development and progression. Altogether, we believe that an enhanced understanding of the DIAPH3 signalosome will facilitate more precise clinical decision-making and the development of novel therapeutics against aggressive cancers.
KeywordsDIAPH3, Actin nucleation factor, PCa metastasis, Therapeutic targeting
Many fundamental processes in eukaryotic cells (e.g., cytokinesis, morphogenesis, endocytosis, and cell motility) necessitate dynamic rearrangement of the cellular cytoskeleton. Specifically, disassembly and assembly of actin filaments are crucial for the generation of cellular invaginations or protrusions required for the corresponding physiological processes [1]. The assembly of actin filaments from actin monomers does not occur spontaneously; rather, it requires specific factors to overcome kinetic barriers associated with actin nucleation. The formin family is defined by the presence of a formin homology 2 (FH2) domain, which is evolutionarily conserved across animal and plant species, and plays an essential role in cytoskeletal remodeling by nucleating and polymerizing linear actin filaments [2].
In humans, the formin family comprises 15 distinct members. Based on phylogenetic analysis of the FH2 domain, formins can be categorized into seven subgroups: diaphanous (Dia), dishevelled associated activator of morphogenesis (DAAM), formin-related gene in leukocytes (FRL), formin homology domain-containing protein (FHOD), inverted formin (INF), formin (FMN), and delphilin [2,3]. Figure1A illustrates the protein structures of these subgroups, with one member from each subgroup as a representative example. The FH2 is the signature domain shared across these subgroups. Diaphanous-related formin 3 (DIAPH3), also referred to as Dia2 or DRF3, is the mammalian homologue of the Drosophila Diaphanous protein. It belongs to the relatively well-studied Dia subgroup, particularly with regard to its structural features. The DIAPH3 gene is located on human chromosome 13q21.2, and its encoded protein plays a crucial role in regulating microtubule and actin cytoskeleton and has been implicated in filopodium [4], plasma membrane bleb [5], invadopodium formation [6], cytokinesis [7], and vesicle transport [8].

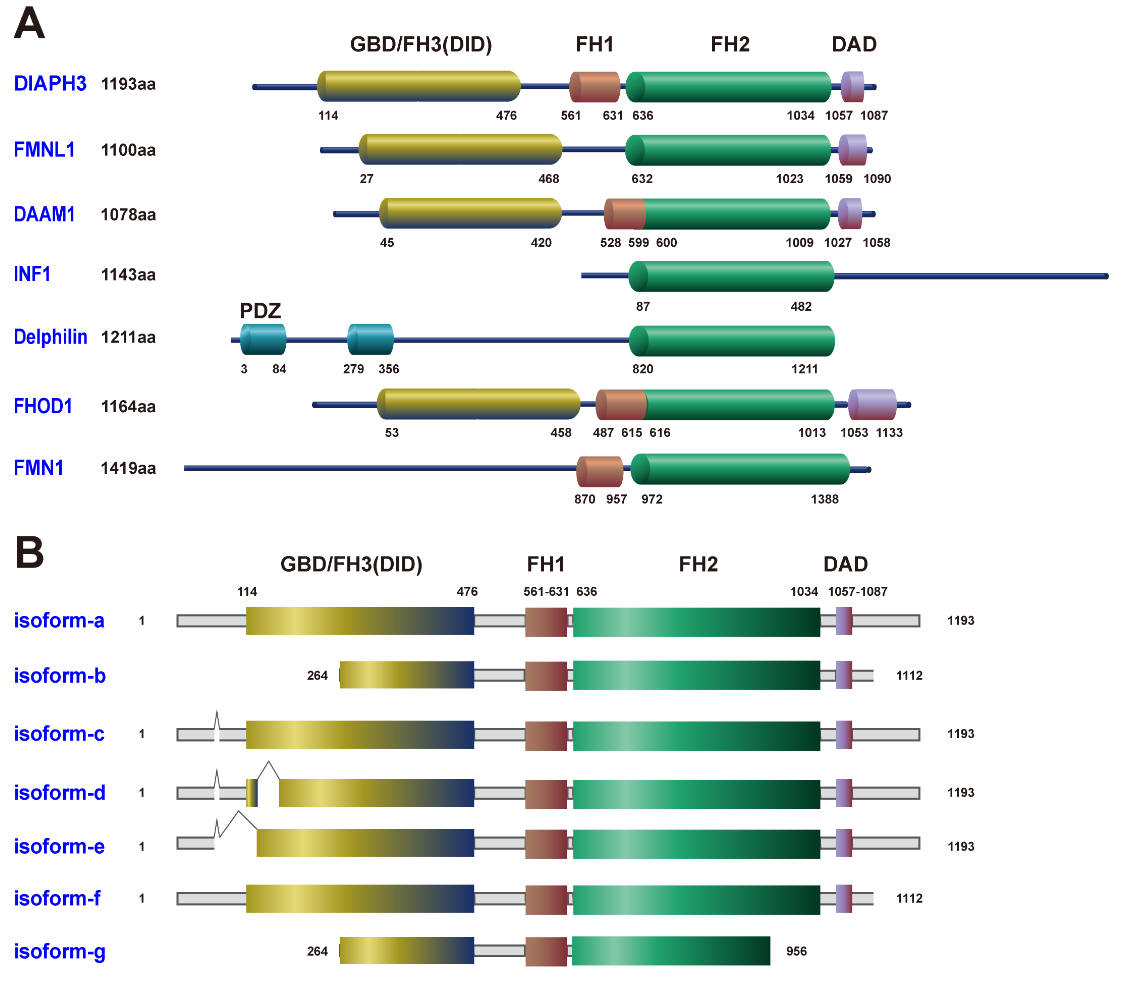
Figure 1 Molecular structures of the formin family. (A) The functional domains of the formin family include the GTPase-binding domain (GBD), diaphanous inhibitory domain (DID), formin homology 1 domain (FH1), formin homology 2 domain (FH2), diaphanous autoinhibitory domain (DAD), and post-synaptic density-95, disks-large and zonula occludens-1 domain (PDZ). Among these, the PDZ domain lacks enzymatic activity but regulates biological processes by facilitating the assembly of protein complexes. (B) The DIAPH3 isoforms generated by alternative splicing in the NCBI and Swiss-Prot databases.
This review focuses on DIAPH3, delving into its cellular structure and the current working models. Furthermore, we discuss its biological roles in various diseases with a particular emphasis on cancer. Of clinical relevance, this review will also signify the potential of DIAPH3 as a prognostic biomarker and a therapeutic target in human cancers.
The full-length DIAPH3 protein features several key structural domains: an N-terminal GTPase-binding domain (GBD, also called FH3), a diaphanous inhibitory domain (DID), a dimerization domain, a coiled-coil region, and two FH domains (FH1 and FH2), followed by a C-terminal diaphanous autoinhibitory domain (DAD) (Figure 1A) [3]. DID is part of the FH3 module. In the prototypical formin structure, the N-terminal DID interacts with the C-terminal DAD, resulting in an auto-inhibitory state [9]. Upon binding to Rho GTPases (such as RhoA, RhoC, Rif, and Cdc42), the GBD alleviates this auto-inhibition, allowing for a conformational change that exposes the functional domains FH1 and FH2. The FH1 domain, located at the N-terminus of the FH2 domain, contains multiple contiguous proline residues that serve as binding sites for the actin monomer binding protein Profilin. By enhancing delivery of new actin monomers to the growing actin filaments, Profilin thereby accelerates filament elongation [10]. Meanwhile, the FH2 domain, consisting of approximately 400 amino acid residues, promotes the incorporation of actin monomers into the barbed end of the growing non-branched actin filament, ultimately enabling the bundling of actin filaments [3]. The mRNA and protein sequences of various DIAPH3 isoforms generated through alternative splicing have been reported in the NCBI and Swiss-Prot databases (Figure 1B). DIAPH3 facilitates isoform-selective actin assembly, which plays distinct and differentially regulated roles during cell adhesion and motility. Notably, isoform-a and isoform-f are the largest and most prevalent isoforms expressed in human tissues, with isoform-f being specifically associated with filopodia formation. In contrast, isoform-b is uniquely involved in the process of plasma membrane blebbing [5]. Such diversity in gene-structure associated functions highlights a complex regulatory mechanism by which DIAPH3 isoforms contribute distinctly to cellular dynamics.
Extracellular cues trigger a reorganization of the cytoskeleton by stimulating cell surface receptors, including G protein-coupled receptors (GPCRs), receptor tyrosine kinases (RTKs), and integrins. For instance, the mitogen lysophosphatidic acid (LPA) activates specific GPCRs, leading to the activation of various guanine nucleotide exchange factors (GEFs) [11]. These GEFs, in turn, regulate the activity of RhoA GTPases. The formation of specific actin-based structures by DIAPH3 is likely dependent on the GTPases and their downstream effectors. For example, the RhoA GTPases Rif and Cdc42 activate DIAPH3 to promote the formation of filopodia. Additionally, other GEFs can activate the Rho GTPases RhoB and RhoD, which localize to the membrane of endosomes, and subsequently stimulate the activation of DIAPH3 [8].
Once the structural auto-inhibition of DIAPH3 is relieved, it fulfills its role in F-actin assembly and participates in various other cellular functions that rely on the actin cytoskeleton (Figure 2A). For instance, DIAPH3 is crucial for E-cadherin dependent adhesion, thereby maintaining the integrity of multicellular epithelial structures [12]. Additionally, DIAPH3 is indispensable for the process of cytokinesis [13]. DIAPH3 localizes to the cytokinetic furrow, where it contributes to the formation of a β-actin network. Alterations in actin isoforms can lead to cytokinetic failure [14]. Moreover, DIAPH3 can undergoes liquid-liquid phase separation (LLPS) to form condensates, resulting in spatial detainment of DIAPH3 from the F-actin assembly sites, thereby inhibiting formation of filopodia under stress [15].
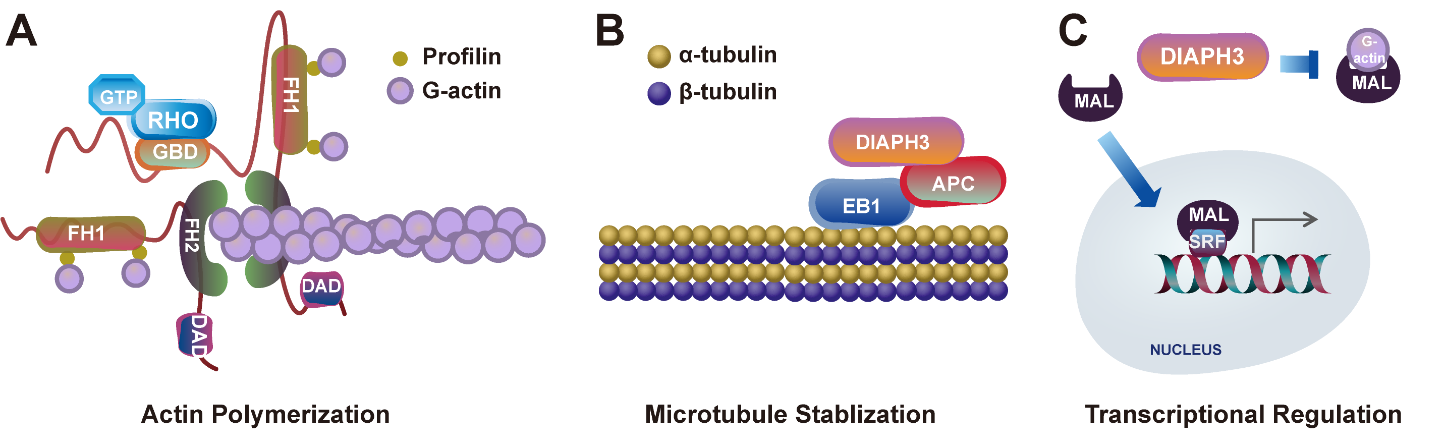
Figure 2 Discovered functions of DIAPH3. (A) DIAPH3 regulates actin nucleation and polymerization. (B) DIAPH3 modulates microtubule dynamics by interacting with microtubule binding proteins. (C) DIAPH3 facilitates the release of megakaryocytic acute leukemia (MAL) into the nucleus, where it acts as a coactivator of serum response factor (SRF) to regulate the transcription of genes related to the cytoskeleton.
Interestingly, DIAPH3 is also implicated in functions that are independent of actin. A construct encompassing the FH1 and FH2 domains of DIAPH3 has been shown to induce stable microtubules without relying on its dimerization and actin nucleation properties [16]. DIAPH3 interacts with the microtubule plus-end binding protein EB1 and adenomatous polyposis coli (APC), facilitating the stabilization of microtubules at the leading edge of the cell during polarized cell migration (Figure 2B) [16]. Notably, downregulation of DIAPH3 leads to a decrease in microtubule stability, which in turn increases susceptibility to taxane drugs that inhibit microtubule dynamics [17]. Furthermore, membrane-derived microvesicles, which are regulated by microtubules, are secreted from prostate cancer (PCa) cells and have been shown to depend on DIAPH3 expression [18]. Moreover, mass spectrometry-based quantitative proteomic analysis has revealed that the interactome of mDia2 (the mouse homolog of DIAPH3) is involved in cell cycle, cell death regulation, and post translational modification, particularly in ubiquitination [19]. DIAPH3 forms a complex with FBXO3 and p53, modulating p53 activity in an actin nucleation-independent and conformation-insensitive manner. These observations raise a possibility that DIAPH3 not only acts as an actin assembly factor but also functions in various other cellular processes [19].
Emerging evidence suggests that DIAPH3 also plays crucial roles in the nucleus. It contains functional nuclear localization sequences, justifying a possible involvement in nuclear activities. Although the specific requirements for DIAPH3’s function within the nuclear environment have yet to be clearly established, recent findings highlight its potential role in transcriptional regulation. Actin polymerization alleviates the inhibition between actin monomers and megakaryocytic acute leukemia (MAL), enabling MAL to translocate into the cell nucleus. As a coactivator of serum response factor (SRF), MAL promotes the transcription of various genes associated with the cytoskeleton, including focal adhesion kinase (FAK), β1-Integrin, Talin, Zyxin, and Vinculin (Figure 2C) [20,21]. Moreover, there is growing interest in the notion that formins may influence transcription by remodeling the nucleoskeleton [22]. A recent study has shown that functional mutations in DAAM2, a member of the formin family, are associated with androgen insensitivity syndrome [23]. DAAM2 is enriched in the nucleus, where its localization correlates with that of the androgen receptor (AR), forming actin-dependent transcriptional droplets in response to androgen dihydrotestosterone (DHT). DAAM2 polymerizes actin directly at the AR to promote droplet coalescence, and nuclear actin polymerization, which is required for prostate specific antigen (PSA) expression in PCa cells [23]. Similar to DAAM2, DIAPH3 can also undergo LLPS to form comparable condensate structures [15]. Based on these findings, it is conceivable that DIAPH3 plays a role in the dynamic reorganization of nuclear structures, thereby contributing to the regulation of gene transcription.
Overall, while the precise mechanisms underlying DIAPH3’s functions in the nucleus remain to be fully elucidated, its potential involvement in these processes opens new avenues for better understanding the role of formin members in cellular dynamics.
Given the intrinsic role of DIAPH3 in regulating actin and microtubule dynamics, which are intricately linked to many other cellular processes, it is expected that dysregulation of DIAPH3 may contribute to a number of pathologies. In patients with autosomal dominant auditory neuropathy type 1 (AUNA1), a heterozygous G-to-A transition has been identified at position 172 in the 5’ -untranslated region (UTR) of the DIAPH3 gene. This mutation leads to a 2- to 3-fold increase in DIAPH3 mRNA levels and a 1.5-fold increase in protein expression [24]. Drosophila models with constitutive overexpression of the mutant DIAPH3 gene in the auditory organ exhibit impaired sound response [24]. Further investigation has identified an additional heterozygous mutation adjacent to the previously reported AUNA1-associated mutation [25]. These findings suggest that AUNA1 is primarily caused by the overexpression of DIAPH3 gene, as a result of a mutation within the transcriptional regulatory site. Consistent with its role in regulating microtubule dynamics, the microtubule meshwork undergoes aberrant targeting to the apical aspect of inner hair cells in DIAPH3 -overexpressing transgenic mice, ultimately leading to a loss of the capability to transmit incoming auditory stimuli [26].
DIAPH3 has also been strongly linked to autism spectrum disorder (ASD), particularly due to the identification of genetic variants in autistic patients. Exome sequencing has revealed a missense mutation in the region of DIAPH3 that encodes FH2 domain, alongside a second mutation in another autism-related gene, SET2 [27]. The disrupted neurogenesis associated with these mutations may contribute to the underlying mechanisms of ASD. Mechanistically, conditional knockout (KO) of DIAPH3 in the cerebral cortex of mice profoundly alters spindle components, disrupts neurogenesis, and leads to cortical malformations and autism-related behaviors [28]. These discoveries underscore the importance of DIAPH3 in neuron- and autism-related diseases. However, there is still much more to be discovered regarding the function of DIAPH3 in other human diseases.
Aberrant organization of the cytoskeleton and the subsequent modulation of downstream signaling events play important roles in tumorigenesis. Specifically, disorganized cytoskeleton, characterized by a decrease in F-actin levels, is a driving force behind the shift of ovarian cancer cells towards a more aggressive and invasive phenotype [29]. Serving as a critical actin nucleator, DIAPH3 is positioned to play a crucial role in regulating the dynamics of actin filament assembly and contributing to the cytoskeletal alterations that propel cancer progression. Therefore, conducting in-depth investigations into the precise functions of DIAPH3 in these processes is imperative for unveiling the underlying mechanisms and pinpointing potential therapeutic targets for cancer treatment development.
To systematically unravel the role and underlying mechanisms of DIAPH3 in cancer, we conducted a comprehensive analysis of the transcriptional and mutational landscape of DIAPH3 across 32 human cancer types through The Cancer Genome Atlas (TCGA) database. Gene expression analysis revealed that DIAPH3 is generally upregulated in most cancers (18 out of 24) when compared to normal counterparts, with the exception of kidney chromophobe carcinoma (KICH), where it is found to be downregulated (Figure 3A). Owing to a lack of expression data in normal tissues, certain cancer types are not included in this analysis. These data indicate an overall trend of upregulation of DIAPH3 in human cancers, suggesting, likely, an oncogenic role for it in tumorigenesis.
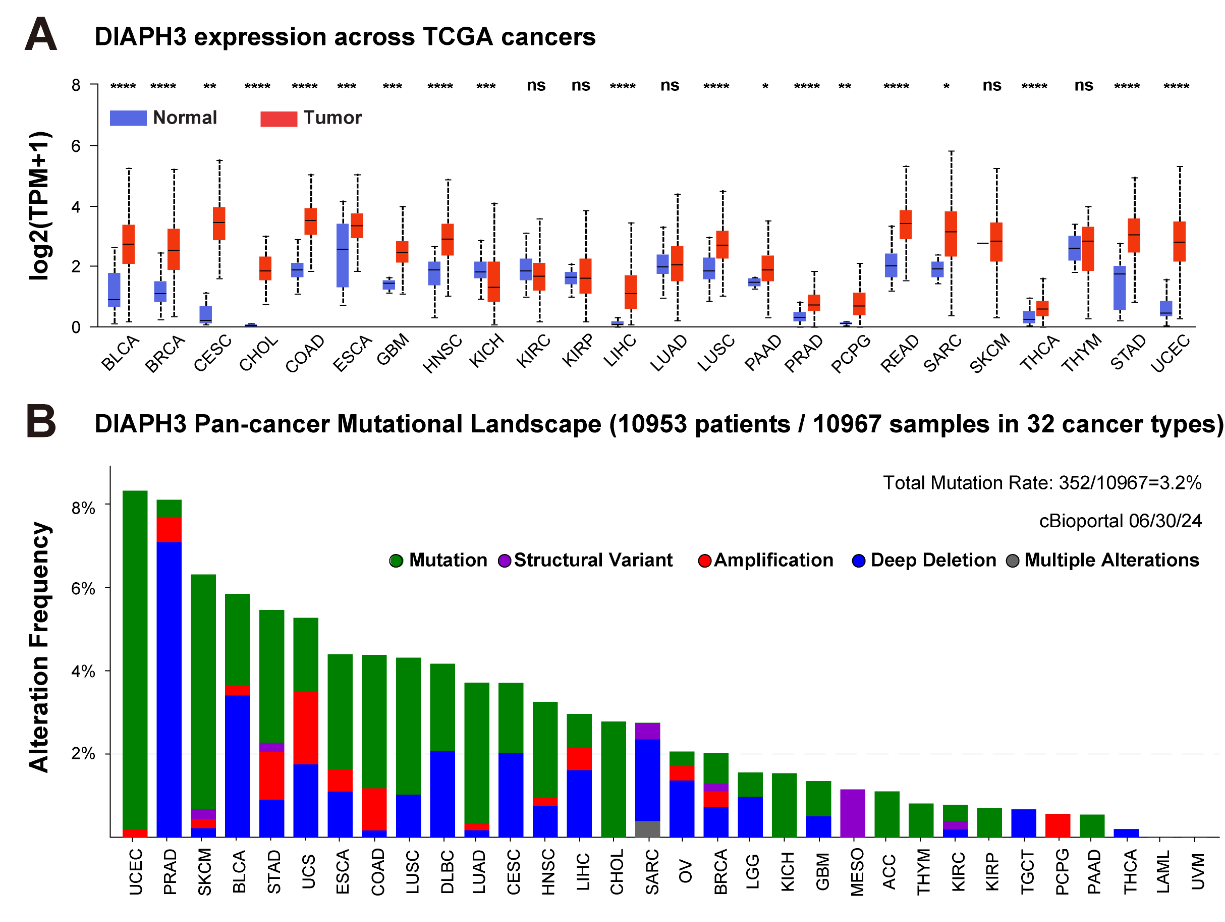
Figure 3 Transcriptomic and mutation landscape of the DIAPH3 gene across 32 human cancers. (A) Pairwise comparison of DIAPH3 expression between normal and tumor tissues in the indicated TCGA cancer types. (B) Bar plots illustrating the cumulative aberration frequencies of DIAPH3 associated with the indicated genetic alterations. Data derived from TCGA pan-cancer analysis encompassing 10,953 patients representing 32 cancer types were viewed through cBioPortal. Adrenocortical Cancer (ACC); Bladder Cancer (BLCA); Breast Cancer (BRCA); Cervical Cancer (CESC); Bile Duct Cancer (CHOL); Colon Cancer (COAD); Diffuse Large B Cell Lymphoma (DLBC); Esophageal Cancer (ESCA); Glioblastoma (GBM); Head and Neck Cancer (HNSC); Kidney Chromophobe Carcinoma (KICH); Kidney Clear Cell Carcinoma (KIRC); Kidney Papillary Cell Carcinoma (KIRP); Acute Myeloid Leukemia (LAML); Lower Grade Glioma (LGG); Liver Cancer (LIHC); Lung Adenocarcinoma (LUAD); Lung Squamous Cell Carcinoma (LUSC); Mesothelioma (MESO); Ovarian Cancer (OV); Pancreatic Adenocarcinoma (PAAD); Pheochromocytoma & Paraganglioma (PCPG); Prostate Adenocarcinoma (PRAD); Rectum Adenocarcinoma (READ); Sarcoma (SARC); Skin Cutaneous Melanoma (SKCM); Stomach Cancer (STAD); Testicular Cancer (TGCT); Thyroid Cancer (THCA); Thymoma (THYM); Uterine Corpus Endometrial Carcinoma(UCEC); Uterine Carcinosarcoma (UCS); Ocular melanomas (UVM).
Genomic alterations are often linked to gene dysregulation. To systematically uncover the underlying mechanisms of DIAPH3 misexpression, we surveyed the mutational landscape of DIAPH3 genes across 32 human cancers. Results identified a collectively low mutation frequency in human cancers, with an average mutation rate of only 3.2% (Figure 3B). This rate varies among different cancer types, ranging from 0.2% in thyroid carcinoma (THCA) to 8.32% in uterine corpus endometrial carcinoma (UCEC). Mutations were identified as a primary form of genomic alterations, particularly in cancers such as UCEC, skin cutaneous melanoma (SKCM), and lung adenocarcinoma (LUAD). This overall very low mutation rate indicates a limited contribution of genomic alteration to the global transcriptomic dysregulation of DIAPH3. Epigenetic modifications may thus play a crucial role in DIAPH3 misexpression. Studies have shown that the super enhancer-regulated long non-coding RNA (LncRNA) LINC01089 is involved in the downregulation of DIAPH3 expression [30]. LINC01089 binds to heterogeneous nuclear ribonucleoprotein M (hnRNPM), thereby enhancing hnRNPM-mediated skipping of DIAPH3 mRNA exon 3. This specific exon 3 contains a significant m6A-modified site that can be recognized by IGF2BP3, consequently promoting DIAPH3 mRNA stability. By skipping exon 3, the expression of DIAPH3 is subsequently reduced [30]. Further studies examining the mechanisms driving DIAPH3 dysregulation are key to dissecting the complex role of DIAPH3 in cancer biology.
In the past few decades, there has been a growing focus on the involvement of DIAPH3 in tumor progression, spanning from primary tumor development to metastasis. DIAPH3 has been reported to promote the proliferation of cervical cancer cells by activating the mTOR signaling pathway [31]. Additionally, overexpression of DIAPH3 in lung cancer enhances tumor growth by weakening the interaction between STK38 and MEKK, resulting in an activation of the mitogen-activated extracellular signal-regulated kinase (MEK)/extracellular regulated protein kinases (ERK) pathway (Figure 4A) [32]. Notably, significant attention has been directed towards its role in cancer cell migration, invasion, and metastasis.
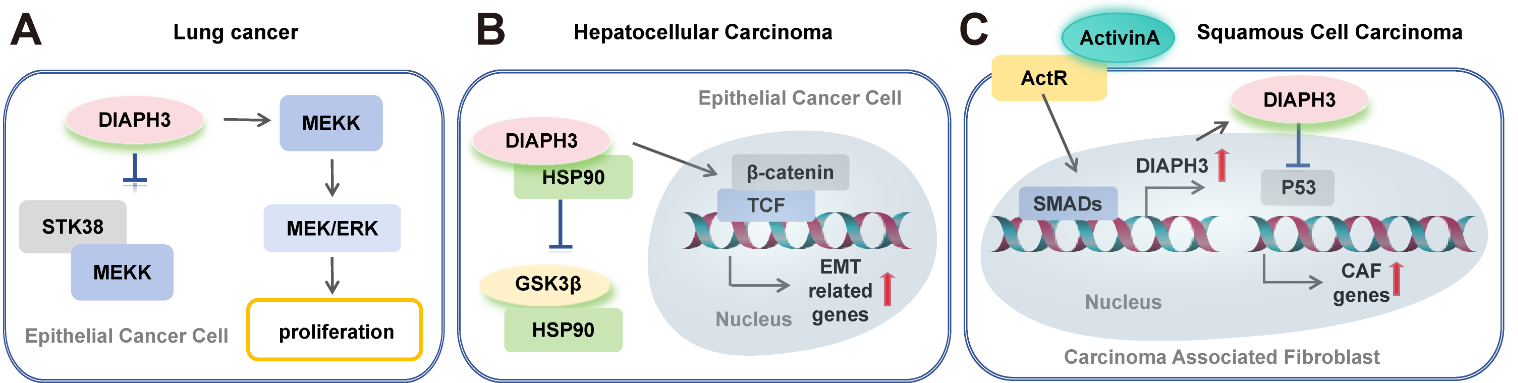
Figure 4 DIAPH3 signalosome in tumor progression. (A) High DIAPH3 expression in lung cancer promotes cancer cell proliferation via the MEK/ERK signaling pathway. (B) Overexpression of DIAPH3 enhances its interaction with HSP90, inhibits the binding of HSP90 to GSK3β, and activates the β-catenin/TCF signaling pathway in hepatocellular carcinoma. (C) Activin A signaling induces the upregulation of DIAPH3, which further contributes to tumor development by reducing the nuclear pool of p53 and promoting the expression of carcinoma-associated fibroblasts (CAFs) marker genes, as well as fibroblast proliferation and migration. Arrows indicate promotion, while perpendicular lines denote inhibitory effects.
Tumor cells possess a dynamic cytoskeleton, enabling them to shift between different motility programs and regulate actin-rich protrusive structures that promote cell motility in response to environmental cues. DIAPH3 is involved in the formation of these actin-rich protrusions, which are essential for migration, such as lamellipodia [33] and filopodia [34]. In cancer, DIAPH3 regulates invasion by facilitating the efficient formation of invadopodia. These actin-based protrusions of the plasma membrane endow cells with matrix proteolytic activity, helping invasive cancer cells to breach the basement membrane (BM) and navigate through the three-dimensional (3D) extracellular environment [35]. In breast cancer (BRCA) cells, silencing DIAPH3 results in a marked reduction of F-actin and cortactin-rich invadopodia, leading to a significant decrease in matrix degradation and, consequently, the invasion capacity of the cancer cells [6].
Hallmark changes associated with the epithelial-mesenchymal transition (EMT) include loss of E-cadherin expression, upregulation of matrix metalloproteases (MMPs), activation of transcriptional regulators, and an adoption of a spindle-like polarized fibroblastic morphology. The EMT program often triggers cancer metastasis. Altered DIAPH3 affects cell morphology and can active EMT. In hepatocellular carcinoma (HCC) cells, highly expressed DIAPH3 promotes the EMT by activating the β-catenin/TCF signaling [36]. After binding with HSP90, DIAPH3 disrupts the interaction between GSK3β and HSP90, resulting in an accumulation of β-catenin and transcriptional activation of several EMT-related genes such as Snail (Figure 4B) [36]. Besides the transition from an adherent, epithelial phenotype to a migratory, mesenchymal phenotype through EMT, cancer cells can also undergo further transformation into an amoeboid phenotype, termed the mesenchymal-to-amoeboid transition (MAT). MAT is characterized by a rounded morphology, extensive actomyosin contractility, and rapid extension and retraction of membrane protrusions, all while exhibiting reduced dependence on proteolysis for migration [37]. DIAPH3 plays a crucial role in regulating of MAT by modifying the microtubule cytoskeleton. Loss of DIAPH3 disrupts microtubule dynamics, impairs endocytosis, and evokes amoeboid properties [38]. Functional inhibition of DIAPH3 through association with its negative regulator, Diaphanous interacting protein (DIP), destabilizes the F-actin cortex, thereby driving membrane blebbing (a hallmark of amoeboid motility) in BRCA cells and promoting tumor cell invasion in 3D matrix [39].
Besides playing critical roles in regulating morphological plasticity during cancer cell motility, studies suggest that DIAPH3 also contributes to cancer progression by modulating the tumor microenvironment (TME). The TME consists of a complex array of components, including tumor cells and stromal cells (such as endothelial cells, tumor-associated macrophages (TAMs), cancer-associated fibroblasts (CAFs), and other immune cells), as well as extracellular matrix (ECM) proteins. These elements form an intricate network that guides tumor initiation, growth, invasion, metastasis, and response to therapies [40]. Among these components, CAFs are particularly crucial as they can secrete a range of growth factors, cytokines, chemokines, proteinases, and ECM proteins to influence tumorigenesis [41]. It is reported that DIAPH3 may contribute to cancer development through CAFs. In the context of BRCA, the activation of YAP leads to the transcriptional upregulation of several cytoskeletal components in CAFs, with one vital regulator being DIAPH3. The cytoskeletal rearrangement modulated by DIAPH3 contributes to matrix stiffening, cancer cell invasion, and angiogenesis [42]. Similar mechanisms have been identified in skin cancer, where activin A (a member of the transforming growth factor-beta (TGF-β) family) exhibits elevated levels in CAFs [43]. Activin A transcriptionally activates DIAPH3 via Smad2, and its interaction with p53 results in the destabilization of nuclear p53, diminishing its transcriptional control. Ultimately, this cascade leads to the generation of a protumorigenic matrisome and secretome (Figure 4C) [43]. On one hand, CAFs actively upregulate their own DIAPH3 to remodel the ECM and promote tumor invasion. On the other hand, these fibroblasts also regulate cancer cell migration and invasion by secreting CXCL12, which modulates the DIAPH3-directed cytoskeleton in BRCA cells [44]. Together, the implication in regulation of morphological plasticity and the TME underscores DIAPH3’s multifaceted roles in cancer biology.
Altered expression of DIAPH3 has been frequently observed in human cancers and has been causally linked to tumorigenesis. Examples of the roles played by DIAPH3 in cancers are summarized in Table 1. While DIAPH3 is often found to be protumorigenic, promoting progression in cancers such as lung [32], breast [42] and pancreatic [45] cancer, there are also instances where it might suppress tumor progression. Particularly, there are reported conflicting roles of DIAPH3 within a specific cancer type. Next, we dissect such situation using PCa as a prime example. An earlier study in 2012 [38] reported that DIAPH3 is downregulated or even completely lost in some samples of prostate, liver, and breast cancers. Further experiments demonstrate that silencing DIAPH3 in Ras-overexpressing mammary epithelial cells (HMEC) and DU145 PCa cells significantly enhances their migratory and invasive capabilities, as well as their potential for distant metastasis. While these results imply a potential tumor suppressor role for DIAPH3, it is crucial to acknowledge the limitations of the study, which utilized a single set of expression data from a PCa cohort with a small sample size (4 normal vs. 16 tumor tissues). Additional data analysis of a different PCa cohort indicated a relationship between the loss of DIAPH3 copy number and a higher Gleason score. However, as previously discussed, the frequency of mutations and deletions involving DIAPH3 in cancers is globally very low, and a reduction in copy number does not always correspond to a decrease in its mRNA levels.
Table 1 DIAPH3 functions in different cancer types.
On the other hand, analysis of patient data from the largest TCGA database reveals a contrasting picture, where DIAPH3 is significantly overexpressed in PCa (Figure 3A). This elevated expression is positively correlated with tumor malignancy and is associated with a poor prognosis for PCa patients [38]. Additionally, some basic bioinformatic analyses of DIAPH3 have been conducted in pan-cancer studies. Specifically, gene enrichment assays in PCa have shown that DIAPH3 expression is negatively correlated with the p53 signaling pathway and tumor inflammation, while positively correlated with tumor proliferation, Myc target genes, angiogenesis, and the TGF-β signaling pathway. Tumor immune estimation resource (TIMER) analysis has also indicated that DIAPH3 expression in PCa is negatively correlated with immune responses and the infiltration of various immune cells, including B cells, natural killer (NK) cells, T effector cells, and macrophages [46]. These findings suggest that DIAPH3 plays an oncogenic role. Such discrepancy highlights a need for detailed studies to elucidate the precise role and mechanism of DIAPH3, in order to devise novel therapeutic targets against aggressive PCa.
Due to its critical roles in cancer, the potential of DIAPH3 as a prognostic biomarker or therapeutic target has been actively pursued. We conducted a systematic LASSO Cox regression analysis on the expression levels of DIAPH3 to construct a prognostic model. Our findings suggest that, in most cases, DIAPH3 is linked to an unfavorable prognosis (Figure 5A), which is consistent with its elevated expression in the majority of cancer types (Figure 3A). Specifically, high levels of DIAPH3 mRNA correlate with poor outcomes in several cancers, including adrenocortical carcinoma (ACC), KICH, pancreatic adenocarcinoma (PAAD), prostate adenocarcinoma (PRAD), and uveal melanoma (UVM). Further Kaplan–Meier (KM) survival analysis demonstrated that an abnormal elevation of DIAPH3 expression in ACC, PAAD, and Lower-Grade Glioma (LGG) serves as a detrimental prognostic factor for cancer patients (Figure 5B). Some studies have also examined the relative expression of DIAPH3 at the mRNA level and/or at the protein level through immunohistochemical analysis of tumor versus normal tissues to identify associations with the clinicopathological characteristics of the cohorts. For example, an abnormal increase in DIAPH3 levels has been observed in cervical cancer, where it has been identified as an unfavorable prognostic factor [46]. In contrast, in glioblastoma, elevated DIAPH3 expression, coupled with an increased proliferation rate, is linked to longer survival, particularly among patients with O6-methylguanine-DNA-methyltransferase (MGMT) methylation [47]. These results underscore the potential of DIAPH3 as a valuable biomarker in cancer prognosis, warranting further investigation into its role in cancer biology.
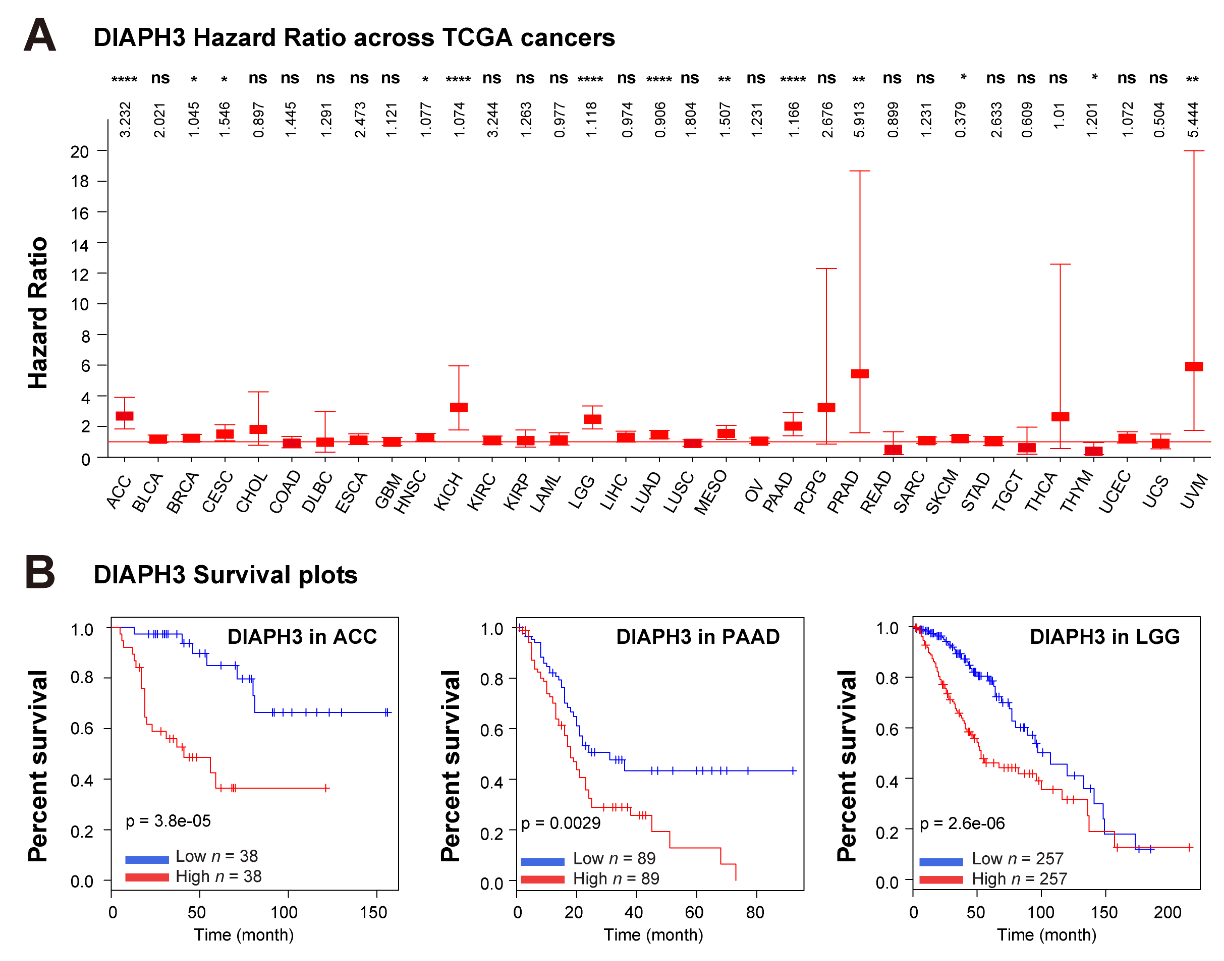
Figure 5 The clinical relevance of DIAPH3. (A) Least absolute shrinkage and selection operator (LASSO) regression analysis of the DIAPH3 gene in 33 human cancers. The hazard ratio of DIAPH3 in each cancer is illustrated in the figure. (B) Kaplan–Meier plots showing DIAPH3 as an unfavorable gene associated with patient overall survival in the indicated cancer types. Data derived from TCGA pan-cancer analysis encompassing 10,953 patients representing 33 cancer types were viewed through cBioPortal. Gene expression and survival analysis were visualized via the online database UALCAN (http://ualcan.path.uab.edu/index.html).
Currently, there are no commercially available DIAPH3-specific inhibitors. However, a small molecular inhibitor of formin FH2 domains, known as SMIFH2, has been identified as an effective inhibitor of formin-mediated actin assembly through a screening of 10,000 commercially available small molecule compounds in vitro [50]. Biochemical analyses indicate that the evolutionarily conserved FH2 domain serves as the molecular target of SMIFH2. Its inhibitory action is specific, affecting both the nucleation and elongation of actin, which are mediated exclusively by formins. Recent studies suggest that SMIFH2 exhibits similar inhibitory effects on six of the seven classes of formins in mammals, including DIAPH3 [51], indicating its broad-spectrum potential against formin-family-mediated actin assembly. In renal epithelial cells, treatment with SMIFH2 resulted in a reduction of filamentous actin [52]. Additionally, in U2OS and HCT116 cells, SMIFH2 treatment disrupted the cycles of actin and microtubule depolymerization and repolymerization, adversely affecting the structure of the Golgi apparatus [53]. The Dia family is widely recognized as a key regulator of the cytoskeleton involved in tumor invasion, and targeting it with SMIFH2 is considered a promising therapeutic approach (Table 2). In experiments with epithelial ovarian cancer cells (SKOV3 and ES2), treatment with SMIFH2 significantly inhibited monolayer cell proliferation. Moreover, combining SMIFH2 with paclitaxel exhibited an additive effect in suppressing the viability of ovarian cancer spheroids [54]. It is worth noting that emerging studies indicate that formins may not be the only target of SMIFH2. Notably, SMIFH2 has been shown to block interferon-induced JAK-STAT signaling [55] and downregulate the p53 protein level in a non-proteasome dependent manner, reducing its transcriptional activity [53]. These findings suggest that caution should be exercised when considering SMIFH2 as an inhibitor of formins due to potential off-target effects.
Table 2 The role of DIAPH3 agonists and antagonists in cancers
In a separate study, compounds IMM-01 and IMM-02 were identified based on their ability to activate the mDia family by disrupting the DID-DAD binding [56]. Molecular docking simulations revealed that they occupy the armadillo-repeat region of the mDia DID structure, a site typically occupied by DAD under physiological conditions [56]. Due to their differing binding orientations, IMM-01 and IMM-02 exhibit varying binding affinities and thus physiological effects. In immortalized NIH-3T3 cells, IMM-01 induced the formation of filamentous lamellipodia and stabilized microtubules, consistent with formin activation, while IMM-02 had a less pronounced effect [56]. This inhibition of microtubule disassembly may contribute to the stabilization of the cytoskeleton and the arrest of mitosis, ultimately leading to the induction of apoptosis, similar to the action of taxane-based chemotherapeutic agents [57]. Notably, continuous treatment with either compound significantly inhibited the invasion of glioblastoma U87 cells, a result achieved through mDia activation [58]. In SW480 colon cancer cells, treatment with 15 μM IMM-01 successfully reversed the persistent activation of the EGFR pathway induced by Dia depletion and inhibited tumor cell proliferation and migration [59]. Additionally, both intraperitoneal and tail vein injections of IMM-01 or IMM-02 were able to inhibit the growth of subcutaneous tumors in nude mice [56]. IMM-01 also acts as an agonist for DAAM2, another member of the formin family containing the DID-DAD self-inhibition structure. It was shown to rescue the impaired formation of lamellipodia caused by DAAM2 knockdown in human podocyte cell lines when treated with 30 μM IMM-01 [60].
Both SMIFH2 and the compounds IMM-01 and IMM-02 show promise as therapeutic agents targeting formins (Table 2). SMIFH2 demonstrates broad-spectrum inhibition of these proteins, while IMM-01 and IMM-02 specifically activate the Dia subgroup of formins, highlighting their potential for targeted cancer therapies. However, these modulators lack specificity, as they can affect the activity of multiple formins. This non-specificity raises the likelihood of side effects, emphasizing a need for search of molecules that can specifically target DIAPH3. In parallel, continued exploration of DIAPH3’s implications in cancer pathogenesis is expected to open new avenues for precision medicine.
DIAPH3 belongs to the formin family and is best known for its essential roles in the nucleation, elongation, and bundling of linear actin filaments. Apart from its functions in actin dynamics, DIAPH3 is also involved in the regulation of microtubules [2], and more broadly, its interactome is associated with cell cycle control, cell death regulation, and post-translational modifications [19]. Interestingly, DIAPH3 may contribute to the dynamic remodeling of the nucleoskeleton, influencing chromatin structure and thus the transcriptional regulation of gene expression. However, the specific function of DIAPH3 within the nuclear environment has not been fully defined.
Dysregulation of DIAPH3 has been associated with several pathologies, including AUNA1 [24] and autism [27]. In particular, its involvement in tumorigenesis has begun to draw global attention. Elevated DIAPH3 expression has not only been observed in many cancer types in the TCGA dataset but has also correlated with poorer clinical outcomes. Studies indicate that DIAPH3 promotes proliferation and tumor growth by activating the mTOR [31] and MEK/ERK [32] signaling pathways. Its crucial role in actin and microtubule dynamics has prompted investigation into its impact on cancer cell migration and invasion. While a few earlier reports linked DIAPH3 deficiency to increased amoeboid cell motility, resulting from reduced microtubule stability [38], a dominant body of evidence supports the notion that DIAPH3 plays an oncogenic role in invasion and metastasis. For instance, DIAPH3 enhances an invasive behavior of BRCA cells by facilitating the actin filament-based formation of invadopodia [6]. Furthermore, DIAPH3 influences cancer progression by remodeling the TME, particularly through interactions with CAFs [42-44].
Controversial results have been reported regarding the roles and significance of DIAPH3 in malignancies. On one hand, high level of DIAPH3 expression has been associated with aggressive cellular behaviors, diminished immune responses, reduced immune cell infiltration, and unfavorable patient outcomes [46]. Conversely, a study has linked elevated DIAPH3 expression to less invasive phenotypes and a more favorable prognosis in prostate cancer [38,46]. This discrepancy may stem from limitations in the bioinformatic analyses with fewer datasets containing a limited number of patients, but it also highlights the possibility that DIAPH3’s role in tumor progression might be context-dependent. As a multifaceted protein, DIAPH3 is believed to be involved in various aspects of tumorigenesis, calling for further research to elucidate its context-specific roles.
Of clinical relevance, we discuss the potential of targeting DIAPH3 for managing human cancers. Experimentally, due to the absence of specific small-molecule DIAPH3 inhibitors, the function of DIAPH3 is evaluated using the broad-spectrum formin inhibitor SMIFH2 [51], along with Dia family agonists [58]. These compounds have yielded promising results in both in vitro and in vivo studies, establishing their potential as targeted cancer therapies. More precisely, future studies aiming at dissecting the underlying mechanisms of DIAPH3 and developing specific DIAPH3 inhibitors are needed to deepen our understanding of formin proteins in cancer.
J.X. and D.Z. conceived the conceptualization, scope, and outline of this review. J.X., L.H., Y.M., D.W. and D.Z. collected and analyzed the relevant references. J.Z. and C.Z. performed the bioinfomatic analysis, J.X., L.H. and D.Z. prepared the schematic illustrations. J.X., L.H., L.Y., Y.F., Y.M., D.W. and D.Z. wrote and revised the manuscript. All authors participated in critical reading and the final approval of the manuscript.
This work was supported by grants from the Shenzhen Science and Technology Program (JCYJ20220530160410024 to DZ and GJHZ20220913144209018 to DW), Guangdong Basic and Applied Basic Research Foundation (2024A1515012312 to DZ), China Postdoctoral Science Foundation (2022M721100 to JX and 2024M750867 to CZ), and the Changsha Natural Science Foundation (KQ2202154 to JX and KQ2208049 to LY).
The material supporting the conclusion of this review has been included within the article.
Not applicable.
Not applicable.
The authors declare that there is no conflict of interests.
The following abbreviations are used in this manuscript:
| 1. | Lappalainen P, Kotila T, Jégou A, Romet-Lemonne G. Biochemical and mechanical regulation of actin dynamics. Nat Rev Mol Cell Bio. 2022;23(12):836-852. [Google Scholar] [CrossRef] |
| 2. | Schönichen A, Geyer M. Fifteen formins for an actin filament: A molecular view on the regulation of human formins. Biochim Biophys Acta Mol Cell Res. 2010;1803(2):152-163. [Google Scholar] [CrossRef] |
| 3. | Breitsprecher D, Goode BL. Formins at a glance. J Cell Sci. 2013;126(1):1-7. [Google Scholar] [CrossRef] |
| 4. | Pellman D, Yang C, Czech L, Gerboth S, Kojima S-i, Scita G, et al. Novel Roles of Formin mDia2 in Lamellipodia and Filopodia Formation in Motile Cells. PLoS Biol. 2007;5(11):e317. [Google Scholar] [CrossRef] |
| 5. | Stastna J, Pan X, Wang H, Kollmannsperger A, Kutscheidt S, Lohmann V, et al. Differing and isoform-specific roles for the formin DIAPH3 in plasma membrane blebbing and filopodia formation. Cell Res. 2011;22(4):728-745. [Google Scholar] [CrossRef] |
| 6. | Lizárraga F, Poincloux R, Romao M, Montagnac G, Le Dez Gl, Bonne I, et al. Diaphanous-Related Formins Are Required for Invadopodia Formation and Invasion of Breast Tumor Cells. Cancer Res. 2009;69(7):2792-2800. [Google Scholar] [CrossRef] |
| 7. | Chen A, Ulloa Severino L, Panagiotou TC, Moraes TF, Yuen DA. Inhibition of polar actin assembly by astral microtubules is required for cytokinesis. Nat Commun. 2021;12(1):2409. [Google Scholar] [CrossRef] |
| 8. | Wallar BJ, DeWard AD, Resau JH, Alberts AS. RhoB and the mammalian Diaphanous-related formin mDia2 in endosome trafficking. Experimental Cell Res. 2007;313(3):560-571. [Google Scholar] [CrossRef] |
| 9. | Alberts AS. Identification of a carboxyl-terminal diaphanous-related formin homology protein autoregulatory domain. J Biol Chem. 2001;276(4):2824-2830. [Google Scholar] [CrossRef] |
| 10. | Paul A, Pollard T. The Role of the FH1 Domain and Profilin in Formin-Mediated Actin-Filament Elongation and Nucleation. Curr Biol. 2008;18(1):9-19. [Google Scholar] [CrossRef] |
| 11. | Young KG, Copeland JW. Formins in cell signaling. Biochim Biophys Acta Mol Cell Res. 2010;1803(2):183-190. [Google Scholar] [CrossRef] |
| 12. | Sahai E, Marshall CJ. ROCK and Dia have opposing effects on adherens junctions downstream of Rho. Nat Cell Biol. 2002;4(6):408-415. [Google Scholar] [CrossRef] |
| 13. | Chen A, Arora PD, McCulloch CA, Wilde A. Cytokinesis requires localized β-actin filament production by an actin isoform specific nucleator. Nat Commun. 2017;8(1):1530. [Google Scholar] [CrossRef] |
| 14. | Shah R, Panagiotou TC, Cole GB, Moraes TF, Lavoie BD, McCulloch CA, et al. The DIAPH3 linker specifies a β-actin network that maintains RhoA and Myosin-II at the cytokinetic furrow. Nat Commun. 2024;15(1):5250. [Google Scholar] [CrossRef] |
| 15. | Zhang K, Huang M, Li A, Wen J, Yan L, Li Y, et al. DIAPH3 condensates formed by liquid-liquid phase separation act as a regulatory hub for stress-induced actin cytoskeleton remodeling. Cell Rep. 2023;42(1):111986. [Google Scholar] [CrossRef] |
| 16. | Bartolini F, Moseley JB, Schmoranzer J, Cassimeris L, Goode BL, Gundersen GG. The formin mDia2 stabilizes microtubules independently of its actin nucleation activity. J Cell Biol. 2008;181(3):523-536. [Google Scholar] [CrossRef] |
| 17. | Morley S, You S, Pollan S, Choi J, Zhou B, Hager MH, et al. Regulation of microtubule dynamics by DIAPH3 influences amoeboid tumor cell mechanics and sensitivity to taxanes. Sci Rep. 2015;5:12136. [Google Scholar] [CrossRef] |
| 18. | Di Vizio D, Kim J, Hager MH, Morello M, Yang W, Lafargue CJ, et al. Oncosome Formation in Prostate Cancer: Association with a Region of Frequent Chromosomal Deletion in Metastatic Disease. Cancer Res. 2009;69(13):5601-5609. [Google Scholar] [CrossRef] |
| 19. | Isogai T, van der Kammen R, Goerdayal SS, Heck AR, Altelaar AFM, Innocenti M. Proteomic Analyses Uncover a New Function and Mode of Action for Mouse Homolog of Diaphanous 2 (mDia2)*. Mol Cell Proteomics. 2015;14(4):1064-1078. [Google Scholar] [CrossRef] |
| 20. | Baarlink C, Wang HC, Grosse R. Nuclear Actin Network Assembly by Formins Regulates the SRF Coactivator MAL. Science. 2013;340(6134):864-867. [Google Scholar] [CrossRef] |
| 21. | Schratt G, Philippar U, Berger J, Schwarz H, Heidenreich O, Nordheim A. Serum response factor is crucial for actin cytoskeletal organization and focal adhesion assembly in embryonic stem cells. J Cell Biol. 2002;156(4):737-750. [Google Scholar] [CrossRef] |
| 22. | Kapoor P, Shen XT. Mechanisms of nuclear actin in chromatin-remodeling complexes. Trends Cell Biol. 2014;24(4):238-246. [Google Scholar] [CrossRef] |
| 23. | Knerr J, Werner R, Schwan C, Wang H, Gebhardt P, Grötsch H, et al. Formin-mediated nuclear actin at androgen receptors promotes transcription. Nature. 2023;617(7961):616-622. [Google Scholar] [CrossRef] |
| 24. | Schoen CJ, Emery SB, Thorne MC, Ammana HR, Śliwerska E, Arnett J, et al. Increased activity of Diaphanous homolog 3 (DIAPH3)/diaphanous causes hearing defects in humans with auditory neuropathy and in Drosophila. P Natl Acad Sci USA. 2010;107(30):13396-13401. [Google Scholar] [CrossRef] |
| 25. | Sánchez-Martínez A, Benito-Orejas JI, Tellería-Orriols JJ, Alonso-Ramos MJ. Neuropatía auditiva autosómica dominante y variante DIAPH3 (c.-173C>T). Acta otorrinolaringologica espanola. 2017;68(3):183-185. [Google Scholar] [CrossRef] |
| 26. | Surel C, Guillet M, Lenoir M, Bourien J, Sendin G, Joly W, et al. Remodeling of the Inner Hair Cell Microtubule Meshwork in a Mouse Model of Auditory Neuropathy AUNA1. eNeuro. 2016;3(6):ENEURO.0295-0216.2016. [Google Scholar] [CrossRef] |
| 27. | Xie J, Li H, Zhu H, Huang L, Li H, Zhang X, et al. Analysis of DIAPH3 gene mutation in a boy with autism spectrum disorder. Zhonghua yi xue yi chuan xue za zhi. 2016;33(4):481-484. (in Chinese). [Google Scholar] [CrossRef] |
| 28. | Lau EOC, Damiani D, Chehade G, Ruiz-Reig N, Saade R, Jossin Y, et al. DIAPH3 deficiency links microtubules to mitotic errors, defective neurogenesis, and brain dysfunction. eLife. 2021;10:e61974. [Google Scholar] [CrossRef] |
| 29. | Creekmore AL, Silkworth WT, Cimini D, Jensen RV, Roberts PC, Schmelz EM. Changes in Gene Expression and Cellular Architecture in an Ovarian Cancer Progression Model. PloS one. 2011;6(3):e17676. [Google Scholar] [CrossRef] |
| 30. | Su T, Zhang NS, Wang T, Zeng JJ, Li WW, Han LY, et al. Super Enhancer-Regulated LncRNA LINC01089 Induces Alternative Splicing of DIAPH3 to Drive Hepatocellular Carcinoma Metastasis. Cancer Res. 2023;83(24):4080-4094. [Google Scholar] [CrossRef] |
| 31. | Wan LL, Zhu JM, Wu QY. Knockdown of DIAPH3 Inhibits the Proliferation of Cervical Cancer Cells through Inactivating mTOR Signaling Pathway. J Oncol. 2021;2021:4228241. [Google Scholar] [CrossRef] |
| 32. | Xiang G, Weiwei H, Erji G, Haitao M. DIAPH3 promotes the tumorigenesis of lung adenocarcinoma. Experimental Cell Res. 2019;385(1):111662. [Google Scholar] [CrossRef] |
| 33. | Gupton SL, Eisenmann K, Alberts AS, Waterman-Storer CM. mDia2 regulates actin and focal adhesion dynamics and organization in the lamella for efficient epithelial cell migration. J Cell Sci. 2007;120(19):3475-3487. [Google Scholar] [CrossRef] |
| 34. | Tominaga T, Sahai E, Chardin P, McCormick F, Courtneidge SA, Alberts AS. Diaphanous-related formins bridge Rho GTPase and Src tyrosine kinase signaling. Mol Cell. 2000;5(1):13-25. [Google Scholar] [CrossRef] |
| 35. | Linder S, Cervero P, Eddy R, Condeelis J. Mechanisms and roles of podosomes and invadopodia. Nat Rev Mol Cell Bio. 2022;24(2):86-106. [Google Scholar] [CrossRef] |
| 36. | Dong L, Li ZJ, Xue LY, Li G, Zhang CY, Cai ZH, et al. DIAPH3 promoted the growth, migration and metastasis of hepatocellular carcinoma cells by activating beta-catenin/TCF signaling. Mol Cell Biochem. 2018;438(1-2):183-190. [Google Scholar] [CrossRef] |
| 37. | Friedl P, Wolf K. Plasticity of cell migration: a multiscale tuning model. J Cell Biol. 2010;188(1):11-19. [Google Scholar] [CrossRef] |
| 38. | Hager MH, Morley S, Bielenberg DR, Gao SZ, Morello M, Holcomb IN, et al. DIAPH3 governs the cellular transition to the amoeboid tumour phenotype. EMBO Mol Med. 2012;4(8):743-760. [Google Scholar] [CrossRef] |
| 39. | Wyse MM, Lei J, Nestor-Kalinoski AL, Eisenmann KM. Dia-Interacting Protein (DIP) Imposes Migratory Plasticity in mDia2-Dependent Tumor Cells in Three-Dimensional Matrices. PloS one. 2012;7(9):e45085. [Google Scholar] [CrossRef] |
| 40. | Jin M-Z, Jin W-L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct Target Ther. 2020;5(1):166. [Google Scholar] [CrossRef] |
| 41. | Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16(9):582-598. [Google Scholar] [CrossRef] |
| 42. | Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol. 2013;15(6):637-646. [Google Scholar] [CrossRef] |
| 43. | Cangkrama M, Wietecha M, Mathis N, Okumura R, Ferrarese L, Al - Nuaimi D, et al. A paracrine activin A–mDia2 axis promotes squamous carcinogenesis via fibroblast reprogramming. EMBO Mol Med. 2020;12(4):e11466. [Google Scholar] [CrossRef] |
| 44. | Aboussekhra A, Dvorak KM, Pettee KM, Rubinic-Minotti K, Su R, Nestor-Kalinoski A, et al. Carcinoma associated fibroblasts (CAFs) promote breast cancer motility by suppressing mammalian Diaphanous-related formin-2 (mDia2). PloS one. 2018;13(3):e0195278. [Google Scholar] [CrossRef] |
| 45. | Rong Y, Gao J, Kuang T, Chen J, Li Ja, Huang Y, et al. DIAPH3 promotes pancreatic cancer progression by activating selenoprotein TrxR1 - mediated antioxidant effects. J Cell Mol Med. 2020;25(4):2163-2175. [Google Scholar] [CrossRef] |
| 46. | Chen X, Xie L, Qiao K, Zhu X, Ren J, Tan Y. The pan-cancer analysis identified DIAPH3 as a diagnostic biomarker of clinical cancer. Aging. 2023;15(3):689-704. [Google Scholar] [CrossRef] |
| 47. | Chehade G, El Hajj N, Aittaleb M, Alkailani MI, Bejaoui Y, Mahdi A, et al. DIAPH3 predicts survival of patients with MGMT-methylated glioblastoma. Front Oncol. 2024;14:1359652. [Google Scholar] [CrossRef] |
| 48. | Wu YF, Xu ZJ, Fu GH, Chen XY, Tian JJ, Cai HR, et al. Identification of a cisplatin resistant-based prognostic immune related gene signature in MIBC. Transl Oncol. 2024;44:101942. [Google Scholar] [CrossRef] |
| 49. | Zhang BX, Hu Q, Li YC, Xu CX, Xie XR, Liu P, et al. Diaphanous-related formin subfamily: Novel prognostic biomarkers and tumor microenvironment regulators for pancreatic adenocarcinoma. Front Mol Biosci. 2022;9:910950. [Google Scholar] [CrossRef] |
| 50. | Rizvi SA, Neidt EM, Cui J, Feiger Z, Skau CT, Gardel ML, et al. Identification and Characterization of a Small Molecule Inhibitor of Formin-Mediated Actin Assembly. Chem Biol. 2009;16(11):1158-1168. [Google Scholar] [CrossRef] |
| 51. | Orman M, Landis M, Oza A, Nambiar D, Gjeci J, Song K, et al. Alterations to the broad-spectrum formin inhibitor SMIFH2 modulate potency but not specificity. Sci Rep. 2022;12(1):13520. [Google Scholar] [CrossRef] |
| 52. | Efremov YM, Dokrunova AA, Efremenko AV, Kirpichnikov MP, Shaitan KV, Sokolova OS. Distinct impact of targeted actin cytoskeleton reorganization on mechanical properties of normal and malignant cells. Biochim Biophys Acta Mol Cell Res. 2015;1853(11):3117-3125. [Google Scholar] [CrossRef] |
| 53. | Isogai T, van der Kammen R, Innocenti M. SMIFH2 has effects on Formins and p53 that perturb the cell cytoskeleton. Sci Rep. 2015;5(1):9802. [Google Scholar] [CrossRef] |
| 54. | Ziske MA, Pettee KM, Khaing M, Rubinic K, Eisenmann KM. SMIFH2-mediated mDia formin functional inhibition potentiates chemotherapeutic targeting of human ovarian cancer spheroids. Biochem Biophys Res Commun. 2016;472(1):33-39. [Google Scholar] [CrossRef] |
| 55. | Thoidingjam LK, Blouin CM, Gaillet C, Brion A, Solier S, Niyomchon S, et al. Small Molecule Inhibitors of Interferon - Induced JAK - STAT Signalling. Angew. Chem. Int. Ed . 2022;61(32):e202205231. [Google Scholar] [CrossRef] |
| 56. | Lash LL, Wallar BJ, Turner JD, Vroegop SM, Kilkuskie RE, Kitchen-Goosen SM, et al. Small-Molecule Intramimics of Formin Autoinhibition: A New Strategy to Target the Cytoskeletal Remodeling Machinery in Cancer Cells. Cancer Res. 2013;73(22):6793-6803. [Google Scholar] [CrossRef] |
| 57. | Fitzpatrick JM, de Wit R. Taxane Mechanisms of Action: Potential Implications for Treatment Sequencing in Metastatic Castration-resistant Prostate Cancer. Eur Urol. 2014;65(6):1198-1204. [Google Scholar] [CrossRef] |
| 58. | Arden JD, Lavik KI, Rubinic KA, Chiaia N, Khuder SA, Howard MJ, et al. Small-molecule agonists of mammalian Diaphanous–related (mDia) formins reveal an effective glioblastoma anti-invasion strategy. Mol Biol Cell. 2015;26(21):3704-3718. [Google Scholar] [CrossRef] |
| 59. | Huang R, Wu C, Wen J, Yu J, Zhu H, Yu J, et al. DIAPH3 is a prognostic biomarker and inhibit colorectal cancer progression through maintaining EGFR degradation. Cancer Med. 2022;11(23):4688-4702. [Google Scholar] [CrossRef] |
| 60. | Schneider R, Deutsch K, Hoeprich GJ, Marquez J, Hermle T, Braun DA, et al. DAAM2 Variants Cause Nephrotic Syndrome via Actin Dysregulation. Am J Hum Genet. 2020;107(6):1113-1128. [Google Scholar] [CrossRef] |
| 61. | Poincloux R, Collin O, Lizárraga F, Romao M, Debray M, Piel M, et al. Contractility of the cell rear drives invasion of breast tumor cells in 3D Matrigel. P Natl Acad Sci USA. 2011;108(5):1943-1948. [Google Scholar] [CrossRef] |
| 62. | Fessenden TB, Beckham Y, Perez-Neut M, Ramirez-San Juan G, Chourasia AH, Macleod KF, et al. Dia1-dependent adhesions are required by epithelial tissues to initiate invasion. J Cell Biol. 2018;217(4):1485-1502. [Google Scholar] [CrossRef] |
| 63. | Rana MK, Aloisio FM, Choi C, Barber DL, Gardel M. Formin-dependent TGF-β signaling for epithelial to mesenchymal transition. MBoC. 2018;29(12):1465-1475. [Google Scholar] [CrossRef] |
| 64. | Liu D, Fu X, Wang Y, Wang X, Wang H, Wen J, et al. Protein diaphanous homolog 1 (Diaph1) promotes myofibroblastic activation of hepatic stellate cells by regulating Rab5a activity and TGFβ receptor endocytosis. FASEB J. 2020;34(6):7345-7359. [Google Scholar] [CrossRef] |

![]()
Copyright © 2026 Pivot Science Publications Corp. - unless otherwise stated | Terms and Conditions | Privacy Policy




