© 2025 by the author(s). This is an Open Access article distributed under the terms of the Creative Commons License Attribution 4.0 International (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly credited.
Abstract
Breast cancer stem cells (BCSCs) are a small proportion of stem-like breast cancer cells with more tumorigenic and metastatic capacity in comparison to the bulk tumor cells, which are capable of self-renewal and generating differentiated cells. BCSCs are resistant to chemotherapy and radiotherapy. More and more research indicate that BCSCs are heterogeneous and have plasticity, which refer to their ability to switch between different subtypes of stem-like cells and differentiated cells. There is some evidence showing that BCSC heterogeneity and plasticity plays a role on therapeutic resistance and metastasis of breast cancer. In this review, we discussed the roles of extrinsic (rewired tumor microenvironment, including hypoxia, microbiota) and intrinsic (pro-tumor signaling) factors on regulating BCSC heterogeneity and plasticity, leading to various malignant behaviors of BCSCs, including epithelial-mesenchymal transition (EMT)/mesenchymal-epithelial transition (MET), immune evasion, vasculogenesis and so on. Moreover, we also discussed the potential therapeutic strategies to target BCSCs.
Keywords
breast cancer stem cell, tumor microenvironment, plasticity, heterogeneity, targeting therapy
1. Introduction
Breast cancer is the most common cancer in women worldwide, and is a leading cause of cancer mortality in women [1]. Treatment of breast cancer is largely based on the expression of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor 2 (HER2/ERBB2). The hormone receptors ER/PR-positive patients account for around 70% of breast cancer patients, who are treated with hormonal therapy [2]. HER2 expression, which is observed in approximately 15–25% of breast cancers, is mainly relevant in the choice of anti-HER2 targeted therapy. Triple-negative breast cancer (TNBC), defined by the absence of all three markers, is the most aggressive subtype of breast cancer due to the lack of targeted therapeutic options. About 15% of breast cancers are TNBC [2]. Chemotherapy and immunotherapy are currently the most commonly used systematic therapies in TNBC patients. However, tumor recurrence, metastasis and treatment resistance remain the leading causes of mortality, particularly in TNBC patients.
Substantial advances have been made in understanding the heterogeneity of breast cancer. Cancer stem cells (CSCs) are a subpopulation of cancer cells that can self-renew and differentiate into different cell lineages, thereby contributing to tumor relapse and treatment resistance. Self-renewal refers to a process that occurs during cell division, whereby at least one daughter cell maintains stem cell-like phenotypes, including long-term proliferative potential [3]. The CSC self-renewal capacity can be assessed by serial passaging of cancer cells in vivo [4]. CSCs have been discovered in many types of cancer, including breast cancer, glioblastoma, colon cancer and prostate cancer [5]. One of the most profound implications of CSCs is their contribution to tumor heterogeneity. Due to their clinical significance, several biomarkers have been identified to characterize CSCs, correlating with diagnosis, therapy sensitivity and prognosis. Dormant CSCs are thought to be more resistant to current therapies [6]. Importantly, CSCs exhibit high plasticity, modifying their phenotypic and functional appearance under different conditions [7]. By understanding the specific markers and signaling pathways associated with CSCs, novel treatments could potentially inhibit their self-renewal or induce differentiation, thus eradicating the seeds of the tumor. Here, we discussed the heterogeneity and plasticity of CSCs in breast cancer and novel treatment strategies to target CSCs.
2. Breast Cancer Stem Cells (BCSCs)
BCSCs are a small proportion of breast cancer cells that are capable of self-renewal and generate differentiated cancer cells [8]. The origin of BCSCs is still unclear. One of the theories is that they derive from mammary stem cells (MaSC), as they share similar markers and a MaSC-enriched population has been observed in preneoplastic tissue of mammary tumor-prone mice [6]. MaSCs possess the ability to differentiate into various mammary cell lineages, contributing to the gland's functional architecture during puberty, pregnancy, and lactation. MaSCs lacks the expression of hormone receptors in mice and they might become BCSCs after gaining the mutation [9]. Further defining the regulation mechanisms of MaSC might help us understanding the origin of BCSCs.
BCSCs have been extensively studied due to their malignant behavior such as tumorigenesis, therapeutic resistance and high metastatic potential. The classic method to identify CSCs involves cell sorting by flow cytometry to isolate cell populations based on specific surface markers, followed by inoculation of cells at limited dilutions in serial xenotransplantation assay to assess the tumor-initiating cell frequency [3]. A number of alternative approaches include three-dimensional (3D) culture systems and single-cell RNA sequencing, etc [3]. The main characteristics of BCSCs are their high heterogeneity and plasticity, which contribute remarkably to their malignancy and pose a challenge for efficient cancer therapeutics. Heterogeneity refers to their subpopulations that express different cellular markers. Plasticity refers to their ability to switch between different states regulated by the tumor microenvironment and pro-tumor signaling pathways.
The mammary gland is a highly regenerative organ that goes through significant changes during the menstrual cycle, pregnancy, lactation, and involution, and stem cells play important roles during these process. How these changes affect BCSCs and breast oncogenesis is still controversial. A previous study showed that pregnancy in the adult mouse does not alter the proportion of mammary epithelial stem/progenitor cells [10]. Epidemiological studies have demonstrated that an early, full-term pregnancy at young age may reduce the lifetime risk of breast cancer [11]. However, pregnancy generally offers protection against the development of ER/PR+ tumors, while the breast cancers that emerge shortly after pregnancy typically do not express these hormone receptors [12]. During pregnancy, the breast undergoes significant morphological and functional changes, primarily driven by a surge in hormones such as estrogen and progesterone. These hormonal shifts stimulate mammary gland development and differentiation, leading to the proliferation of epithelial cells and increased stromal support. The microenvironment with growth factors and cytokines promotes the survival and expansion of BCSCs [13]. Furthermore, another study revealed that parity reduced mammary repopulating activity but did not affect mammary stem cells defined as CD24+ CD29/CD49fhi in mice [14].
3. BCSC Heterogeneity and Plasticity
A number of subpopulations within breast cancer have been identified to possess high tumor-initiating potential. The most commonly used markers to identify BCSCs include CD24– CD44+, ALDH, and CD49f [15,16].
The initial BCSC population to be identified was that of CD24–/low CD44+ cells [4]. These cancer cells demonstrate a substantial capacity for tumor initiation. As few as 100 breast cancer cells with EpCAM+, high levels of CD44, and low levels of CD24 expression were able to efficiently form tumors when inoculated in immune-deficient mice [4]. The tumorigenic subpopulation could be serially passaged. Each time, these cells generated new tumors containing additional CD44+CD24–/low Lineage– tumorigenic cells as well as the phenotypically diverse mixed populations of non-tumorigenic cells present in the initial tumor [4]. CD44 is a cell-surface glycoprotein that binds hyaluronic acid (HA) and many other extracellular matrix components [17]. CD44 is involved in cell-cell interactions, cell adhesion and migration. Alternative splicing of CD44 could determine breast cancer stem cell state [18]. CD44 standard splice isoform (CD44s) is the predominant isoform expressed in BCSCs and positively associates with the CSC gene signatures. In contrast, the CD44 variant splice isoforms (CD44v) exhibit an inverse association with the CSC signatures [18]. Mounting evidence have suggested that CD44 functions not only as CSC markers but also as critical regulators of cancer stemness, including self-renewal, tumor initiation, and metastasis [17].
Another frequently used BCSCs marker is aldehyde dehydrogenase 1 (ALDH1). ALDH1 is a member of the aldehyde dehydrogenase protein family, which catalyzes the oxidation of intracellular aldehydes. The measurement of ALDH1 activity via the Aldefluor assay is a frequently employed method for the analysis of BCSCs in both cell lines and animal models. ALDH1 plays a role in the early differentiation of BCSCs through oxidizing retinol to retinoic acid [16]. The activity of ALDH1A1 in BCSCs has been demonstrated to remodel myeloid-derived suppressor cells, thereby promoting breast cancer progression [19].
CD49f, also known as integrin-α6, is a further BCSC marker, as well as a common CSC marker [20]. It forms homodimers with β4-integrin, thereby facilitating binding to laminin and enhancing epithelial cell adhesion to the extracellular matrix [21]. CSCs, which are enriched in the CD24+ CD29+/CD49f+ cell population from a Brca1-mutant mouse model, exhibited a markedly elevated migration capacity relative to CD24– CD29–/CD49f– cells [22]. The dynamics of the CD49f+ tumor-initiating population is associated with taxane resistance in TNBC [23].
Notably, CD24–/low CD44+ mesenchymal-like BCSCs are primarily quiescent and localized at the tumor invasive front, whereas ALDH+ epithelial-like BCSCs, are proliferative, and are located more centrally [24]. Although breast cancer cells with both CD24–/low CD44+ and ALDH+ expression represent a rare population, they have been observed to exhibit the greatest tumorigenic and invasive capacity [24]. Importantly, a recent study has demonstrated that ALDH1A3 is the key regulator that determines the balance of ALDH+ and CD24–CD44+ breast cancer stem cells [25]. While ALDH1A3 increases the number of ALDH+ breast cancer cells, it inversely suppresses the CD24– CD44+ population by retinoic acid signaling. It can therefore be concluded that BCSCs constitute a heterogeneous population, which is regulated by a number of different factors.
The abundance of BCSCs in different breast cancer subtypes has also been explored. The tumors enriched with CD44+ CD24– or ALDH+ subpopulation were more correlated with basal-like and HER2-positive breast cancers [26,27]. In line with these results, BCSCs were rare in ER+ PR+ luminal subtype breast cancers [28]. Moreover, within a tumor, CD44+ CD24− cells showed lower ER and PR expression compared to CD44− CD24+ cells [28]. These results may also explain the poor prognosis of TNBC and HER2-positive patients.
The causes of BCSC heterogeneity is complicated and not yet fully elucidated. One theory is the occurrence of series of mutations in differentiated somatic cells. A key aspect of this theory is the concept of de-differentiation of differentiated cell, allowing them to be reverted to a more stem-like state under certain conditions [29]. This plasticity may underlie the heterogeneity observed in BCSCs, as different differentiated cells may respond uniquely to mutagenic stress, leading to diverse tumor phenotypes and behaviors. In contrast, the second theory proposes the occurrence of mutations in MaSC or progenitors [30]. This theory suggests that BCSCs arise from mutations occurring in the normal mammary stem cell compartment or its immediate progenitors. These mutations can lead to the dysregulation of key pathways involved in stem cell maintenance and differentiation, resulting in the heterogeneity and plasticity of BCSCs. However, sequencing analysis revealed that the majority of somatic mutations are shared between CSCs and bulk tumors [31]. Therefore, future efforts are still needed to deeply understand the causes of BCSC heterogeneity and plasticity.
4. Extrinsic Factors Regulating BCSCs Heterogeneity and Plasticity
The heterogeneity of BCSCs is typically attributed to their inherent plasticity. This plasticity refers to their capacity to transition between stem-like and differentiated states [32]. Multiple mechanisms have been identified as regulators of BCSCs plasticity and heterogeneity. Here we discuss how the tumor microenvironment components, hypoxia, tumor-resident microbiota, and tumor cell intrinsic signaling pathways influence the heterogeneity and plasticity of BCSCs.
4.1 Tumor ecosystem
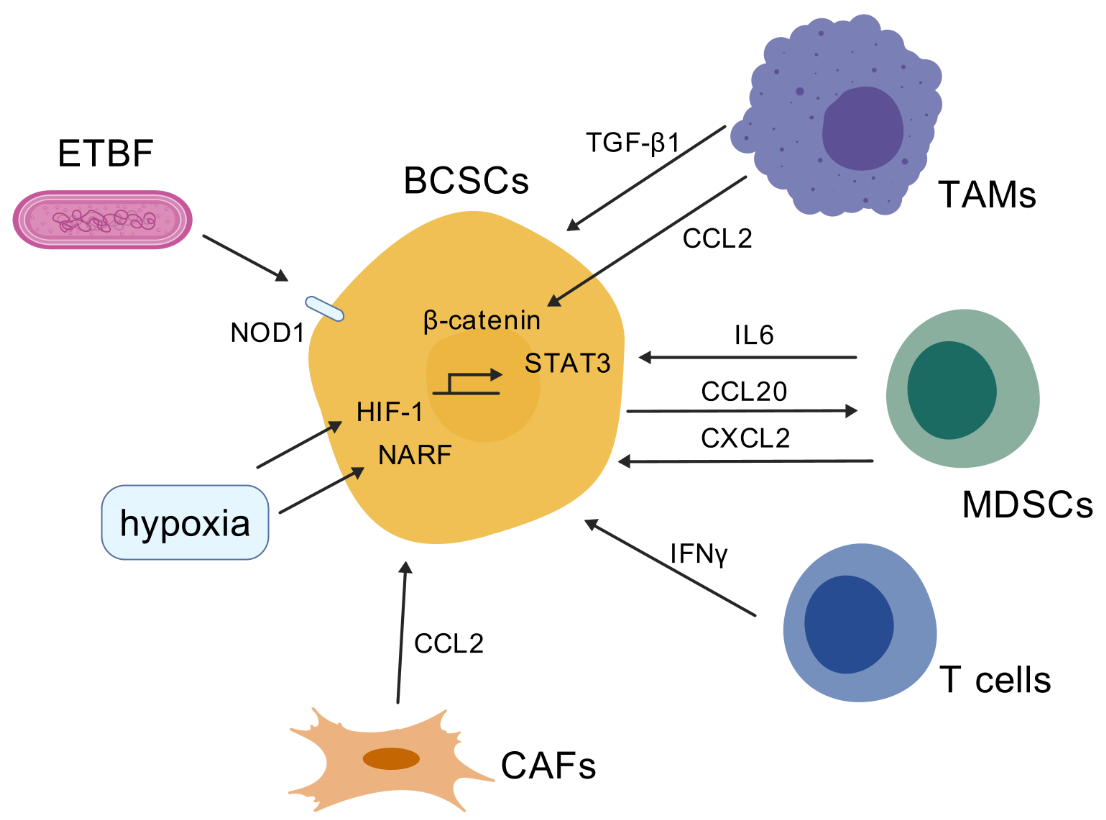
Breast cancer consists of multiple cell types, including a large variety of immune and other host cells, which create the tumor microenvironment (TME). The crosstalk between all these cellular and structural components is essential for breast cancer growth and progression by accumulating phenotypic and genetic heterogeneity. The tumor microenvironment has been identified as a driving force behind BCSC heterogeneity and plasticity, with interactions between immune cells, stromal cells, and the extracellular matrix influencing BCSC expansion. The immune cells in the TME regulate BCSC plasticity through the establishment of positive feedback loops [33]. Here, we summarize ways in which the various cell types that comprise the TME, including macrophages, myeloid-derived suppressor cells, T cells and cancer-associated fibroblasts, contribute to the BCSC stemness and plasticity. The mechanisms are summarized in Figure 1 (created with BioGDP.com [34]).
4.1.1 Immune cells
Macrophage
Tumor-associated macrophages (TAMs) constitute the most abundant immune population in the breast tumor microenvironment. TAMs have been demonstrated to facilitate cancer cell proliferation and metastasis, while also exerting immunosuppressive effects on the adaptive immune cells that populate the TME. Macrophages that have been activated are typically classified into two categories: M1-like macrophages, which are primarily involved in pro-inflammatory responses, and M2-like macrophages, which are primarily involved in anti-inflammatory responses.
Recent studies have demonstrated that TAMs promote epithelial-mesenchymal transition (EMT) and the cancer stem cell properties in breast cancer via the CCL2/AKT/β-catenin signaling [35,36]. In addition, Wang et al. recently reported that Rab13, a small GTPase, is highly expressed in BCSCs. Rab13 was found to control the membrane translocation of C-X-C chemokine receptor type 1/2 (CXCR1/2), allowing tumor cells to interact with TAMs and cancer-associated fibroblasts, and establish a supportive BCSC niche [37]. Furthermore, macrophage-induced ERK-TGF-β1 signaling in MCF7 breast cancer cells result in cancer stem cell plasticity and EMT which are completely reversible [38]. Additionally, M2-like TAMs contribute to increase cancer cell stemness in TNBC cells via secreting VEGFA, which were evaluated with stem cell markers, including CD24, CD44, OCT-4, Nanog and SOX-2. Reciprocally, elevated VEGFA expression by TAM-educated TNBC cells acts as a regulator of macrophage polarization [39]. In light of these findings, it can be concluded that TAMs play a pivotal role in regulating BCSC stemness and plasticity.
Myeloid-derived suppressor cell(MDSC)
Myeloid-derived suppressor cells (MDSCs) are activated neutrophils and monocytes with potent immunosuppressive activity. Two distinct subtypes of MDSCs have been identified: monocytic myeloid-derived suppressor cells (M-MDSCs) and granulocytic polymorphonuclear MDSCs (PMN-MDSCs). M-MDSCs show a stronger immunosuppressive effect [33]. Nonetheless, PMN-MDSCs play an important role in regulating tumor featured immune responses [40].
MDSC contribute to the development of stem-like phenotypes in breast cancer cells, which in turn facilitate immune suppression and tumor recurrence. For example, MDSCs promoted tumor growth by enhancing sphere formation and increasing the percentage of ALDH+ BCSCs in breast cancer as well as by suppressing T-cell activation. These effects were mediated by the cross-talk between the IL6-STAT3 and NOTCH pathways in cancer cells with MDSC [41]. MDSC was found to be correlated with the presence of BCSCs in breast cancer samples and was also identified as an independent predictor of poor survival outcomes [41]. Additionally, it was demonstrated that PMN-MDSCs, which are modulated by CCL20 from cancer cells, can increase ALDH+ BCSCs and stemness genes (eg. KLF4, NANOG, SOX9, etc.) via the CXCL2-CXCR2 pathway in breast cancer [42].
T cell
Although T cells are known for their tumor-killing ability, recent studies reveal that ineffective immune responses not only fail to clear a tumor, but also promote stemness and tumor-forming capacity in cancer cells. Interferon gamma (IFNγ) produced by activated T cells following immunotherapy directly converts non-CSCs to CSCs [43]. IFNγ enhances several CSC phenotypes, including resistance to chemo- and radiotherapy, as well as metastasis formation [43]. Cognate interaction with nonlytic CD8+T cells increased the proportion of CD44+CD24– BCSCs in a cell-to-cell contact- or proximity-dependent manner [44].
4.1.2 Cancer-Associated Fibroblast (CAF)
CAFs play a pivotal role in the reactive stroma within the TME, as they not only interact with cancer cells extensively via secreted cytokines or cell–cell adhesion but also indirectly influence cancer cells via remodeling of the extracellular matrix and immune cell infiltration [45]. For instance, primary CAFs activated by cocultured breast cancer cells produce higher levels of chemokine CCL2, which induces the CSC phenotype in breast cancer [46]. Increased CCL2 expression in activated fibroblasts required STAT3 activation by diverse breast cancer-secreted cytokines, and in turn, induced the mammosphere formation and self-renewal of expansion in breast cancer cells via NOTCH1, constituting a cancer-stroma-cancer signaling circuit [46]. In addition, Su et al. have found that CD10+GPR77+ CAFs provide a survival niche for CSCs, thereby promoting tumor formation and chemoresistance [47]. Mechanistically, CD10+GPR77+ CAFs are driven by persistent NF-κB activation via p65 phosphorylation and acetylation, which is maintained by complement signaling via GPR77. In line with this notion, targeting these CAFs with a neutralizing anti-GPR77 antibody abolishes tumor formation and restores chemosensitivity [47]. On the other hand, BCSCs reciprocally promote the formation of CAFs. Breast cancer stem cells are found to upregulate IRF6 in stromal fibroblasts to induce stromagenesis, in order to sustain the BCSC tumor niche [48].
4.1.3 Hypoxia
Hypoxia is a key feature of the breast cancer TME that promotes expression of the transcription factor hypoxia-inducible factor 1 (HIF-1) and is correlated with unfavorable prognosis and chemoresistance. HIF-1 induces the expression or activity of stem cell pluripotency factors, including NANOG, KLF4, OCT4 and SOX2 [49,50]. It is also noteworthy that HIF-1 facilitates the evasion of phagocytosis and the maintenance of BCSCs by activating the transcription of CD47 [51]. CD47 functions as a mediator of immune evasion by delivering a 'don't eat me' signal to macrophages [52]. Hypoxia-induced secretion regulates the dissemination of CSCs, mediated by JAK-STAT signaling [53]. A recent study found that nuclear prelamin A recognition factor (NARF) acts as a hypoxia-induced coactivator for OCT4-mediated BCSC specification [49]. It was also illustrated that HIF-2α, but not HIF-1α, induces breast cancer cell stemness under hypoxia [54]. Mechanistically, hypoxia inducible factor-2α (HIF-2α) activates superoxide dismutase 2 (SOD2) under hypoxic conditions, thereby decreasing mitochondrial reactive oxygen species (mtROS) levels. The reduced mtROS transported to the endoplasmic reticulum subsequently activate unfolded protein response (UPR) via GRP78, which eventually confer chemoresistance of cancer cells, increase of OCT4 expression, and both CD44+CD24−BCSC and ALDH1+ BCSC populations [54]. Accordingly, depletion or inhibition of HIF-2α was observed to abolish the hypoxia-induced sphere-forming ability and the percentage of CD44+CD24− cell subpopulation [54]. Of note, glycolysis was found to reprogram BCSC under hypoxic conditions. Pyruvate dehydrogenase kinase 1 (PDK1), a key glycolysis gene, was found to be overexpressed in BCSCs and accumulated in hypoxic regions. The inhibition of PDK1 led to a remarkable reduction in the percentage of ALDH+ BCSC subpopulation, expression of stemness-related transcriptional factor, and sphere-forming ability and tumor growth in breast cancer [55]. Taken together, hypoxia has been identified as one of the critical factors in the TME that sustain the BCSC stemness.
4.1.4 Tumor-resident microbiota
The roles of microbiota for cancer cell stemness, metastasis and response to treatment remain poorly understood, having only recently emerged as a topic of investigation. Tumor-resident intracellular microbiota, albeit at low biomass, were shown to promote metastatic colonization in breast cancer [56]. It is noteworthy that microbiota enterotoxigenic Bacteroides fragilis (ETBF) was found to be highly enriched in tumors of breast cancer patients who did not respond to taxane-based neoadjuvant chemotherapy [57]. Mechanistically, the toxic protein BFT-1 secreted by ETBF, directly bound to the cytosolic pattern recognition receptor NOD1. NOD1 increased BCSCs by activating the NOTCH1-HEY1 signaling pathway. In line with this notion, NOD1 inhibition and ETBF clearance was shown to increase the chemosensitivity of breast cancer by impairing BCSCs [57]. However, evidence on how the tumor-resident microbiota regulate breast cancer stemness and plasticity is still rare, which warrants further investigation.
4.2 Intrinsic signaling in tumor cells
4.2.1 Notch signaling
It is well established that Notch signaling plays a pivotal role in sustaining the self-renewal and tumor-initiation capacities of BCSCs. The Notch pathway involves four Notch receptors (Notch1-4) and five ligands (Jagged1, Jagged2, Delta-like1 (Dll1), Dll3, and Dll4). The association between NOTCH3 expression and tumor metastasis in ERα+ and TNBC models has been identified [58]. The abrogation of NOTCH3 expression was found to significantly reduce the self-renewal and invasive capacity of ex vivo breast cancer cells, restoring a luminal CD44low/CD24high/ERαhigh phenotype [58]. Additionally, NOTCH4 has been shown to enhance tumorigenesis in TNBC cells via upregulating Nanog expression [59]. NOTCH4 also maintains quiescent mesenchymal-like BCSCs in TNBC cells via activating SLUG and GAS1 [60]. The Notch ligand Dll1 is also a key factor in tumor growth and metastasis and promotes stemness of breast cancer cells [61]. Activation of the NF-κB pathway occurs downstream of Dll1 and is associated with chemoresistance. In addition, a recent study has demonstrated that the tumor suppressor FERM domain-containing protein 3 (FRMD3) functions as an endogenous activator of the Notch signaling pathway, facilitating the basal-to-luminal transformation in mammary epithelial cells [62].
4.2.2 Wnt/β-catenin signaling
The Wnt/β-catenin signaling plays critical roles in the proliferation, migration, and treatment resistance of BCSCs. β-catenin is a key downstream effector of the Wnt signaling pathway. Following the binding of Wnt family proteins to the Frizzled receptor, β-catenin translocates to the nucleus where it interacts with LEF1 and TCF1 to activate canonical targets including CD44, cyclin D1, c-Jun, c-Myc, Met, and MMP-7 [63]. Notably, a recently identified unique subcluster of Socs3highCD11b–CD27– immature NK cells in TNBC samples expressed a reduced cytotoxic granzyme signature and, in mice, were responsible for activating cancer stem cells through Wnt signaling. The activation of BCSCs by NK cells subsequently enhanced tumor progression in mice, whereas depletion of NK cells or Wnt ligand secretion from NK cells resulted in a decrease in tumor progression [64]. In addition, FBXW7 blocks the activation of the Wnt/β-catenin signaling pathway to inhibit the percentage of ALDH+ and CD44+CD24– BCSCs in TNBC by degrading CHD4 protein through ubiquitination [65]. Furthermore, the secretome of BCSCs has been shown to contain MIF, which plays a crucial role in potentiating glycolysis and the subsequent evasion of immune responses. MIF was shown to increase c-MYC-mediated transcriptional upregulation of the glycolytic enzyme aldolase C by activating WNT/β-catenin signaling [66]. Therefore, the Wnt/β-catenin signaling serves as a promising target to inhibit BCSCs.
4.2.3 Hedgehog (Hh) signaling
The Hedgehog (Hh) signaling pathway plays critical roles in regulating cell fate and cell growth during the embryonic development. It is recognized as a key player in tumor growth, stemness and metastasis. Three canonical Hh ligands have been identified: Sonic hedgehog (SHH), Indian hedgehog (IHH), and Desert hedgehog (DHH) [67]. Notably, TSPAN8 has been demonstrated to promote BCSC stemness through activating Sonic Hedgehog signaling [68]. Moreover, the Hedgehog signaling mediates the interaction between BCSCs and cancer-associated fibroblasts (CAF). CSCs secrete the Hedgehog ligand SHH, which regulates CAFs via paracrine activation of Hedgehog signaling. Conversely, CAFs secrete factors that facilitate the expansion and self-renewal of CSCs [69]. The administration of the Hedgehog inhibitor vismodegib decreases CAF and CSC expansion, consequently leading to a reduction in tumor formation. Similarly, in mouse models of TNBC, the Hedgehog ligand produced by neoplastic cells reprograms CAFs to provide a supportive niche for the acquisition of a chemo-resistant, CSC phenotype via FGF5 expression and production of fibrillar collagen [70]. In line with this notion, stromal treatment of patient-derived xenografts with smoothened inhibitors (SMOi) downregulates CSC marker expression and sensitizes tumors to docetaxel. In addition, the Hh signaling also regulates aerobic glycolysis, which is critical for sustaining cancer cell stemness. For instance, ETV4, a key transcription factor in regulating glycolytic gene expression, was shown to promote BCSC stemness by activating CXCR4-mediated sonic Hedgehog signaling [71].
5. Impact of BCSC Heterogeneity and Plasticity
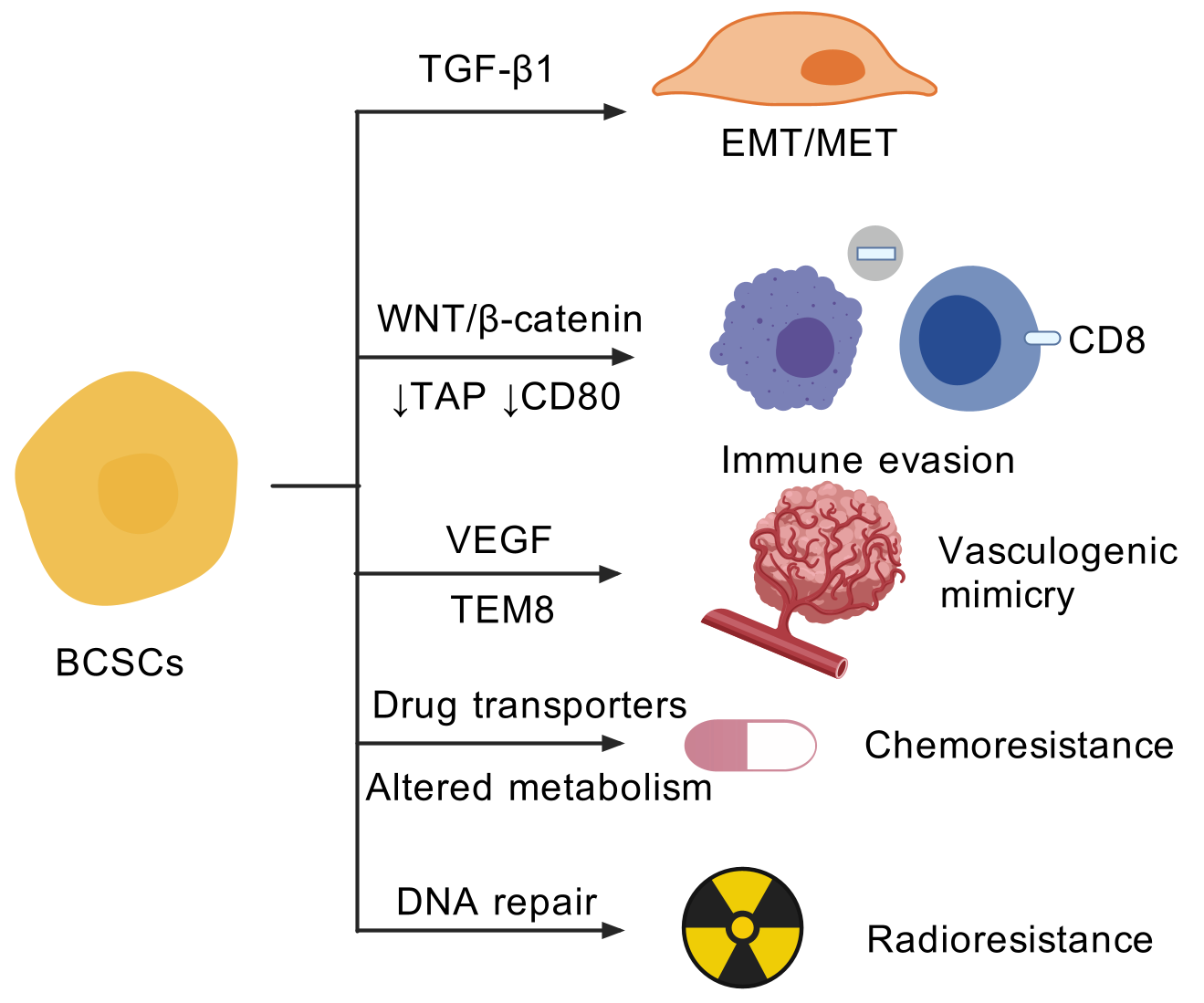
The heterogeneity and plasticity of BCSCs contribute remarkably to their high malignancy. BCSCs are able to switch between different states in response to corresponding signals and environmental stimuli. The consequences of such plasticity include increased invasive capacity via epithelial-mesenchymal transition, immune evasion, vasculogenic mimicry and resistance to chemotherapy and radiotherapy (Figure 2).
5.1 Epithelial-mesenchymal transition (EMT) and Mesenchymal-epithelial transition (MET)
BCSCs display plasticity that enables them to flexibly transit between EMT and MET states. The EMT and MET processes play crucial roles in the metastasis of cancer. EMT is a cell process in which epithelial cells undergo a reversible phenotype change marked by the loss of adhesion ability and acquisition of mesenchymal features. The process is regulated by multiple transcription factors, including ZEB1, Snail and Twist [72]. Tumor cells that have undergone an EMT-like process are more likely to enter the circulation through intravasation and disseminate to distant organs. Conversely, the MET state allows tumor cells to reside in metastatic sites. These phenotypic changes are induced by a wide variety of extracellular signals. Notably, mesenchymal-like BCSCs are characterized as CD24-CD44+, while epithelial-like BCSCs are ALDH+. The gene-expression profiles of mesenchymal-like and epithelial-like BCSCs resemble those of distinct normal basal and luminal stem cells and are remarkably similar across different molecular subtypes of breast cancer [24]. In addition, the BCSCs isolated from primary TNBC specimens express epithelial and mesenchymal markers concomitantly [73]. BCSCs are susceptible to the EMT inducer TGF-β1, leading to the increase of mesenchymal genes and enhanced cell migration [73].
5.2 Immune evasion
BCSCs contribute to the immune evasion phenotype, which may influence the efficacy of immunotherapy. The mechanisms include a reduction in neo-antigens, antigen presentation and crosstalk with immune cells. For instance, radioresistant BCSCs have been observed to enhance DNA double-strand break repair and replication fork protection ability to immunogenic cytosolic DNA, thereby inhibiting the intracellular immune response [74]. Of note, ALDH+ cells in breast cancer, but not CD44+CD24– cells, have reduced expression of transporter associated with antigen processing (TAP) genes and co-stimulatory molecule CD80, which would decrease susceptibility to T cells [75]. Moreover, the upregulated glycolysis in BCSCs cells, which is mediated by WNT/β-catenin signaling suppresses intratumoral cytolytic CD8+ T cells and proinflammatory macrophages while increasing regulatory T cells and tumor-associated neutrophils in the TME [66].
5.3 Vasculogenic mimicry (VM)
The rapid growth of cancer requires an efficient blood vessel network to provide enough oxygen and nutrition. VM elucidates the formation of fluid-conducting channels by highly invasive and genetically unstable tumor cells, thereby contributing to tumor metastasis and treatment resistance [76,77]. The primary pathway involved in tumor angiogenesis is the VEGF pathway. In addition, alternative neovasculogenic pathways play critical roles in tumor progression, as they facilitate the formation of a perfusable vasculogenic-like network, known as vasculogenic mimicry (VM) [78]. Recent studies revealed the associated between CD133+ and USP44+ cancer stem cells and vasculogenic mimicry in breast cancer [79,80]. Of note, tumor endothelial marker 8 (TEM8) has been identified as a marker for VM-forming breast tumor-initiating cells [78]. TEM8 increased active RhoC level and induced phosphorylation of SMAD5, in a cascade essential for promoting stemness and VM capacity of breast cancer cells [78]. Moreover, evidence indicates that endoplasmic reticulum stress was shown to inhibit Matrigel-induced vasculogenic mimicry of breast cancer cells [81]. In light of these findings, it seems reasonable to suggest that targeting the vasculogenic capacity of BCSCs may prove an effective strategy for the treatment of breast cancer.
5.4 Chemoresistance and radioresistance
More and more research shows that BCSCs are that they are resistant to chemotherapies and radiotherapy. Besides the mechanisms mentioned above, other factors also contribute to the treatment resistance. In terms of chemoresistance, CSCs often exhibit upregulation of ATP-binding cassette (ABC) transporters, such as ABCG2 and MDR1 [82,83]. These efflux pumps actively extrude chemotherapeutic agents from the cells, reducing drug accumulation and efficacy. Moreover, BCSCs can enter a quiescent state, remaining in a non-dividing phase of the cell cycle [84]. The dormancy provides a protective mechanism against agents such as chemotherapies that target rapidly dividing cells, rendering conventional chemotherapies ineffective. Furthermore, BCSCs can utilize distinct metabolic pathways to survive under low nutrient conditions and evade the cytotoxic effects of chemotherapies. For instance, fatty acid β-oxidation is found to be critical for BCSC self-renewal and chemoresistance [85]. Regarding to radioresistance, BCSCs possess enhanced DNA repair mechanisms, including upregulated expression of repair proteins, enabling them to efficiently repair the DNA damage induced by radiation, thereby surviving from the treatment [86].
6. Preclinical/Clinical Therapeutic Strategies Targeting BCSCs
Given the pivotal roles that BCSCs play in tumorigenesis and treatment resistance, many therapeutic approaches have been tested in preclinical settings alone or in combination with chemotherapy. A summary of recent novel preclinical therapeutics, along with their respective targets and effects on BCSCs, is presented in Table 1.
The first strategy involves the development of small molecule inhibitors targeting BCSC signalings or the TME. For instance, targeting the Hh signaling pathway may prove an effective strategy to overcome BCSC plasticity. In the phase I clinical trial EDALINE, three of the 12 patients with metastatic TNBC derived clinical benefit from combination therapy with the SMO inhibitor Sonidegib and docetaxel chemotherapy, with one patient experiencing a complete response [70]. In accordance with this hypothesis, novel combinations of JAK2 inhibitors (ruxolitinib and pacritinib) with SMO inhibitors (vismodegib and sonidegib) synergistically suppressed breast cancer stem cells, tumor growth and metastasis [96]. Additionally, CD73 expression is required for the maintenance of CD44+CD24– BCSCs. Quercetin and luteolin, which are commonly used as over-the-counter supplements, combined with paclitaxel could effectively downregulate paclitaxel-enhanced CD73 and CSC-promoting pathways YAP and Wnt. The triple-drug combination could inhibit paclitaxel-enriched CSCs, suppress the growth of TNBC cell lines and patient-derived xenograft organotypic cultures [88]. The FDA-approved PI3 kinase p110а inhibitor, alpelisib (BYL-719), has the potential to inhibit the stemness and EMT phenotype in BCSCs in 3D cultures. A comparison of eribulin-resistant breast cancer cell lines revealed that BYL-719 effectively overcame drug resistance [87].
In addition to small molecule inhibitors, the use of currently approved antibody also showed promising effects. Notably, pharmacological inhibition of IL-6 by tocilizumab potentiates cisplatin cytotoxicity and targets cancer stem cells in TNBC [92]. New antibodies are also in development. The antibody targeting GD2 (Dinutuximab), which is expressed in around 60% of primary TNBC, is able to inhibit TNBC growth by targeting GD2+ breast cancer stem-like cells [93].
The implementation of immune cytotherapy also showed promising results. For instance, DLL4-targeted CAR-T therapy has been shown to eliminate cancer stem cells and reshape immune microenvironment in HER2+ breast cancer, thereby sensitizing neoadjuvant chemotherapy [94]. Likewise, the TEM8 CAR T cells targeted breast cancer stem-like cells, offsetting the formation of mammospheres [95].
Besides preclinical studies, multiple therapeutic agents targeting BCSC in combination with chemotherapy have been tested in clinical trials. Relevant studies have been summarized in Table 2. The metabolism of CSCs in hypoxia suggests an altered balance between mitochondrial oxidative phosphorylation and glycolysis [97]. In line with this notion, specific drugs targeting mitochondrial metabolism, leading to increased ROS levels, may inhibit CSCs. Many classes of FDA-approved antibiotics, including doxycycline, actually target mitochondria. A recent clinical trial (NCT06452394) aims to check for the clinical efficacy of doxycycline to target the BCSCs and improve the response to neoadjuvant chemotherapy in ER+/HER2– breast cancers.
Moreover, cirmtuzumab is a receptor-tyrosine-kinase like orphan receptor 1 (ROR1) antibody [98]. A phase 1b study has evaluated the safety and efficacy of cirmtuzumab combined with paclitaxel for metastatic or locally advanced, unresectable breast cancer (NCT02776917). The stem cell makers, ALDH and CD133, from primary pre-treatment and post-treatment tumor specimens were measured to assess the effects on BCSCs. Furthermore, CXCR1 has been identified as a druggable target on BCSCs. The CXCR1 inhibitor, reparixin, in combination with paclitaxel has been tested in TNBC patients. Unfortunately, the primary endpoint of prolonged PFS was not met [99].
7. Concluding Remarks and Future Perspectives
Overall, the studies on the heterogeneity and plasticity of BCSCs underscores the complexity of these cells and their roles in tumorigenesis, tumor progression and treatment resistance. Accumulating evidence have proved that a number of factors contribute to the plasticity of BCSCs. These include pro-tumor signaling, immune cells and stromal cells in the TME, hypoxia, non-coding RNAs (e.g., miRNAs, and lncRNAs) and the recently discovered tumor-resident microbiota. A comprehensive understanding of the molecular mechanisms and environmental factors that drive BCSC plasticity is essential for the development of targeted therapies and the improvement of patient outcomes. A disruption of the tumor-TME network may prove an effective strategy for the treatment of breast cancer.
Despite fundamental achievements, several questions still remain elusive. First of all, the eradication of BCSCs may prove challenging due to their inherent heterogeneity. The manner in which distinct subtypes of BCSCs compete or collaborate at various stages of tumor progression remains to be elucidated. Recent advances in spatial transcriptomics and single-cell sequencing may facilitate the resolution of these questions. Secondly, it is crucial to choose the optimal treatment regimens of combining BCSC-targeted therapy with systemic therapy, while minimizing the incidence of adverse effects. Thirdly, the majority of BCSC-targeted therapies have been evaluated in preclinical models. Further clinical trials are required to validate the efficacy and safety of these treatments in patients with breast cancer.
Declarations
Ethics Statement
Not applicable.
Consent for Publication
Not applicable.
Funding
This work was supported by The National Key Research and Development Program of China (2023YFC2506400); National Natural Science Foundation of China (W2431054, 82230103, 81930075); “Ten Thousand Plan” - National High-Level Talents Special Support Plan (WR-YK5202101); The Shanghai Pujiang Program (24PJA019).
Competing Interests
The authors have declared that no competing interests exist.
Table 1. Preclinical therapeutics targeting BCSCs.
| Name |
Target |
Effects |
Reference |
| Small molecule inhibitor |
|
|
|
| Sonidegib |
SMO inhibitor |
Sensitizes tumors to docetaxel, improve survival and reduce metastatic burden |
[70] |
| Alpelisib |
PI3 kinase p110а |
Inhibits BCSCs stemness and EMT |
[87] |
| Quercetin/Luteolin |
CD73 |
Inhibits cancer stem cells and increases lymphocyte infiltration |
[88] |
| Ropivacaine |
AKT |
Suppresses stem cells-like properties of breast cancer cells |
[89] |
| FTY720 |
SphK1 |
Inhibit BCSC proliferation |
[90] |
| HDAC3 inhibitor |
HDAC3 |
Suppresses the CSC subpopulation of TNBC |
[91] |
| Antibody-based therapy |
|
|
|
| Tocilizumab |
IL6 |
Potentiates cisplatin cytotoxicity in TNBC |
[92] |
| Dinutuximab |
GD2 |
Inhibits TNBC growth by targeting GD2+ breast cancer stem-like cells |
[93] |
| CAR-T therapy |
|
|
|
| DLL4-targeted CAR-T cells |
DLL4 |
Eliminates CSCs and reshape immune microenvironment in HER2+ breast cancer |
[94] |
| TEM8-targeted CAR-T cells |
TEM8 |
Kills tumor endothelial cells and TEM8+ TNBC cells; offsetts the formation of mammospheres |
[95] |
Table 2. Clinical trials targeting BCSCs.
| Trial NCT Number |
Type |
Name of Intervention Agent |
Drug Target or Biomarker |
Breast Cancer Subtypes |
Number of Patients Enrolled/ to be Enrolled |
| NCT01190345 |
Phase II |
Bevacizumab |
VEGF |
Not specified |
75 |
| NCT06452394 |
Phase II |
Doxycycline |
A broad-spectrum of bacteria |
ER+/HER2- |
50 |
| NCT02776917 |
Phase Ib |
Cirmtuzumab |
ROR1 |
HER2- |
22 |
| NCT02254005 |
Phase I |
Bivatuzumab Mertansine |
CD44v6 |
Metastatic Breast Cancer |
24 |
| NCT06331169 |
Phase Ib |
Anlotinib |
VEGFR/ FGFR/ PDGFR/ c-kit |
HER2-Low Advanced and/or Metastatic Breast Cancer |
42 |
| NCT05550415 |
Phase II |
Simvastatin |
Vimentin/ HMGCR |
TNBC |
26 |
| NCT00645333 |
Phase I/II |
MK-0752 |
Notch |
Advanced or Metastatic Breast Cancer |
30 |
References
| 1. |
Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. CA Cancer J Clin. 2024;74(1):12-49.
[Google Scholar]
[CrossRef]
|
| 2. |
Nolan E, Lindeman GJ, Visvader JE. Deciphering breast cancer: from biology to the clinic. Cell. 2023;186(8):1708-1728.
[Google Scholar]
[CrossRef]
|
| 3. |
Loh JJ, Ma S. Hallmarks of cancer stemness. Cell Stem Cell. 2024;31(5):617-639.
[Google Scholar]
[CrossRef]
|
| 4. |
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983-3988.
[Google Scholar]
[CrossRef]
|
| 5. |
Tang DG. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012;22(3):457-472.
[Google Scholar]
[CrossRef]
|
| 6. |
Lindeman GJ, Visvader JE. Insights into the cell of origin in breast cancer and breast cancer stem cells. Asia Pac J Clin Oncol. 2010;6(2):89-97.
[Google Scholar]
[CrossRef]
|
| 7. |
Wu M, Zhang X, Zhang W, Chiou YS, Qian W, Liu X, et al. Cancer stem cell regulated phenotypic plasticity protects metastasized cancer cells from ferroptosis. Nat Commun. 2022;13(1):1371.
[Google Scholar]
[CrossRef]
|
| 8. |
Zhang R, Tu J, Liu S. Novel molecular regulators of breast cancer stem cell plasticity and heterogeneity. Semin. Cancer Biol. 2022;82:11-25.
[Google Scholar]
[CrossRef]
|
| 9. |
Asselin-Labat ML, Shackleton M, Stingl J, Vaillant F, Forrest NC, Eaves CJ, et al. Steroid hormone receptor status of mouse mammary stem cells. J Natl Cancer Inst. 2006;98(14):1011-1014.
[Google Scholar]
[CrossRef]
|
| 10. |
Britt KL, Kendrick H, Regan JL, Molyneux G, Magnay FA, Ashworth A, et al. Pregnancy in the mature adult mouse does not alter the proportion of mammary epithelial stem/progenitor cells. Breast Cancer Res. 2009;11(2):R20.
[Google Scholar]
[CrossRef]
|
| 11. |
Russo J, Moral R, Balogh GA, Mailo D, Russo IH. The protective role of pregnancy in breast cancer. Breast Cancer Res. 2005;7(3):131-142.
[Google Scholar]
[CrossRef]
|
| 12. |
Britt K, Ashworth A, Smalley M. Pregnancy and the risk of breast cancer. Endocr Relat Cancer. 2007;14(4):907-933.
[Google Scholar]
[CrossRef]
|
| 13. |
Tiede B, Kang Y. From milk to malignancy: the role of mammary stem cells in development, pregnancy and breast cancer. Cell Res. 2011;21(2):245-257.
[Google Scholar]
[CrossRef]
|
| 14. |
Dall GV, Vieusseux J, Seyed-Razavi Y, Godde N, Ludford-Menting M, Russell SM, et al. Parity reduces mammary repopulating activity but does not affect mammary stem cells defined as CD24 + CD29/CD49fhi in mice. Breast Cancer Res Treat. 2020;183(3):565-575.
[Google Scholar]
[CrossRef]
|
| 15. |
Zhang L, Chen W, Liu S, Chen C. Targeting Breast Cancer Stem Cells. Int. J. Biol. Sci. 2023;19(2):552-570.
[Google Scholar]
[CrossRef]
|
| 16. |
Sousa B, Ribeiro AS, Paredes J. Heterogeneity and Plasticity of Breast Cancer Stem Cells. Adv Exp Med Biol. 2019;1139:83-103.
[Google Scholar]
[CrossRef]
|
| 17. |
Wang L, Zuo X, Xie K, Wei D. The Role of CD44 and Cancer Stem Cells. Methods Mol. Biol. 2018;1692:31-42.
[Google Scholar]
[CrossRef]
|
| 18. |
Zhang H, Brown RL, Wei Y, Zhao P, Liu S, Liu X, et al. CD44 splice isoform switching determines breast cancer stem cell state. Genes Dev. 2019;33(3–4):166-179.
[Google Scholar]
[CrossRef]
|
| 19. |
Liu C, Qiang J, Deng Q, Xia J, Deng L, Zhou L, et al. ALDH1A1 Activity in Tumor-Initiating Cells Remodels Myeloid-Derived Suppressor Cells to Promote Breast Cancer Progression. Cancer Res. 2021;81(23):5919-5934.
[Google Scholar]
[CrossRef]
|
| 20. |
Bhat-Nakshatri P, Kumar B, Simpson E, Ludwig KK, Cox ML, Gao H, et al. Breast Cancer Cell Detection and Characterization from Breast Milk-Derived Cells. Cancer Res. 2020;80(21):4828-4839.
[Google Scholar]
[CrossRef]
|
| 21. |
Radisky D, Muschler J, Bissell MJ. Order and disorder: the role of extracellular matrix in epithelial cancer. Cancer Invest. 2002;20(1):139-153.
[Google Scholar]
[CrossRef]
|
| 22. |
Vassilopoulos A, Chisholm C, Lahusen T, Zheng H, Deng CX. A critical role of CD29 and CD49f in mediating metastasis for cancer-initiating cells isolated from a Brca1-associated mouse model of breast cancer. Oncogene. 2014;33(47):5477-5482.
[Google Scholar]
[CrossRef]
|
| 23. |
Gómez-Miragaya J, Palafox M, Paré L, Yoldi G, Ferrer I, Vila S, et al. Resistance to Taxanes in Triple-Negative Breast Cancer Associates with the Dynamics of a CD49f+ Tumor-Initiating Population. Stem Cell Rep. 2017;8(5):1392-1407.
[Google Scholar]
[CrossRef]
|
| 24. |
Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014;2(1):78-91.
[Google Scholar]
[CrossRef]
|
| 25. |
Fernando W, Cruickshank BM, Arun RP, MacLean MR, Cahill HF, Morales-Quintanilla F, et al. ALDH1A3 is the switch that determines the balance of ALDH(+) and CD24(-)CD44(+) cancer stem cells, EMT-MET, and glucose metabolism in breast cancer. Oncogene. 2024;43(43):3151-3169.
[Google Scholar]
[CrossRef]
|
| 26. |
Nakshatri H, Srour EF, Badve S. Breast cancer stem cells and intrinsic subtypes: controversies rage on. Curr Stem Cell Res Ther. 2009;4(1):50-60.
[Google Scholar]
[CrossRef]
|
| 27. |
Honeth G, Bendahl PO, Ringnér M, Saal LH, Gruvberger-Saal SK, Lövgren K, et al. The CD44+/CD24- phenotype is enriched in basal-like breast tumors. Breast Cancer Res. 2008;10(3):R53.
[Google Scholar]
[CrossRef]
|
| 28. |
Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11(3):259-273.
[Google Scholar]
[CrossRef]
|
| 29. |
Badve S, Nakshatri H. Breast-cancer stem cells-beyond semantics. Lancet Oncol. 2012;13(1):e43-8.
[Google Scholar]
[CrossRef]
|
| 30. |
Zhang Z, Christin JR, Wang C, Ge K, Oktay MH, Guo W. Mammary-Stem-Cell-Based Somatic Mouse Models Reveal Breast Cancer Drivers Causing Cell Fate Dysregulation. Cell Rep. 2016;16(12):3146-3156.
[Google Scholar]
[CrossRef]
|
| 31. |
Klevebring D, Rosin G, Ma R, Lindberg J, Czene K, Kere J, et al. Sequencing of breast cancer stem cell populations indicates a dynamic conversion between differentiation states in vivo. Breast Cancer Res. 2014;16(4):R72.
[Google Scholar]
[CrossRef]
|
| 32. |
Saw PE, Liu Q, Wong PP, Song E. Cancer stem cell mimicry for immune evasion and therapeutic resistance. Cell Stem Cell. 2024;31(8):1101-1112.
[Google Scholar]
[CrossRef]
|
| 33. |
Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. 2021;21(8):485-498.
[Google Scholar]
[CrossRef]
|
| 34. |
Jiang S, Li H, Zhang L, Mu W, Zhang Y, Chen T, et al. Generic Diagramming Platform (GDP): a comprehensive database of high-quality biomedical graphics. Nucleic Acids Res. 2025;53(D1):D1670-D1676.
[Google Scholar]
[CrossRef]
|
| 35. |
Chen X, Yang M, Yin J, Li P, Zeng S, Zheng G, et al. Tumor-associated macrophages promote epithelial-mesenchymal transition and the cancer stem cell properties in triple-negative breast cancer through CCL2/AKT/β-catenin signaling. J Cell Commun Signal. 2022;20(1):92.
[Google Scholar]
[CrossRef]
|
| 36. |
Zhang F, Li P, Liu S, Yang M, Zeng S, Deng J, et al. β-Catenin-CCL2 feedback loop mediates crosstalk between cancer cells and macrophages that regulates breast cancer stem cells. Oncogene. 2021;40(39):5854-5865.
[Google Scholar]
[CrossRef]
|
| 37. |
Wang H, Xu H, Chen W, Cheng M, Zou L, Yang Q, et al. Rab13 Sustains Breast Cancer Stem Cells by Supporting Tumor-Stroma Cross-talk. Cancer Res. 2022;82(11):2124-2140.
[Google Scholar]
[CrossRef]
|
| 38. |
Kundu P, Shankar BS. Macrophage induced ERK-TGF-β1 signaling in MCF7 breast cancer cells result in reversible cancer stem cell plasticity and epithelial mesenchymal transition. Biochim Biophys Acta Gen Subj. 2022;1866(11):130215.
[Google Scholar]
[CrossRef]
|
| 39. |
Wang L, Zhang L, Zhao L, Shao S, Ning Q, Jing X, et al. VEGFA/NRP-1/GAPVD1 axis promotes progression and cancer stemness of triple-negative breast cancer by enhancing tumor cell-macrophage crosstalk. Int J Biol Sci. 2024;20(2):446-463.
[Google Scholar]
[CrossRef]
|
| 40. |
Hegde S, Leader AM, Merad M. MDSC: Markers, development, states, and unaddressed complexity. Immunity. 2021;54(5):875-884.
[Google Scholar]
[CrossRef]
|
| 41. |
Peng D, Tanikawa T, Li W, Zhao L, Vatan L, Szeliga W, et al. Myeloid-Derived Suppressor Cells Endow Stem-like Qualities to Breast Cancer Cells through IL6/STAT3 and NO/NOTCH Cross-talk Signaling. Cancer Res. 2016;76(11):3156-3165.
[Google Scholar]
[CrossRef]
|
| 42. |
Zhang R, Dong M, Tu J, Li F, Deng Q, Xu J, et al. PMN-MDSCs modulated by CCL20 from cancer cells promoted breast cancer cell stemness through CXCL2-CXCR2 pathway. Signal Transduct Target Ther. 2023;8(1):97.
[Google Scholar]
[CrossRef]
|
| 43. |
Beziaud L, Young CM, Alonso AM, Norkin M, Minafra AR, Huelsken J. IFNγ-induced stem-like state of cancer cells as a driver of metastatic progression following immunotherapy. Cell Stem Cell. 2023;30(6):818-831.e6.
[Google Scholar]
[CrossRef]
|
| 44. |
Stein RG, Ebert S, Schlahsa L, Scholz CJ, Braun M, Hauck P, et al. Cognate Nonlytic Interactions between CD8(+) T Cells and Breast Cancer Cells Induce Cancer Stem Cell-like Properties. Cancer Res. 2019;79(7):1507-1519.
[Google Scholar]
[CrossRef]
|
| 45. |
Yang D, Liu J, Qian H, Zhuang Q. Cancer-associated fibroblasts: from basic science to anticancer therapy. Exp Mol Med. 2023;55(7):1322-1332.
[Google Scholar]
[CrossRef]
|
| 46. |
Tsuyada A, Chow A, Wu J, Somlo G, Chu P, Loera S, et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. 2012;72(11):2768-2779.
[Google Scholar]
[CrossRef]
|
| 47. |
Su S, Chen J, Yao H, Liu J, Yu S, Lao L, et al. CD10(+)GPR77(+) Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell. 2018;172(4):841-856.e16.
[Google Scholar]
[CrossRef]
|
| 48. |
Muralidharan H, Hansen T, Steinle A, Schumacher D, Stickeler E, Maurer J. Breast Cancer Stem Cells Upregulate IRF6 in Stromal Fibroblasts to Induce Stromagenesis. Cells. 2024;13(17):1466.
[Google Scholar]
[CrossRef]
|
| 49. |
Yang Y, Chen C, Zuo Q, Lu H, Salman S, Lyu Y, et al. NARF is a hypoxia-induced coactivator for OCT4-mediated breast cancer stem cell specification. Sci Adv. 2022;8(49):eabo5000.
[Google Scholar]
[CrossRef]
|
| 50. |
Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc Natl Acad Sci U S A. 2016;113(14):E2047-E2056.
[Google Scholar]
[CrossRef]
|
| 51. |
Zhang H, Lu H, Xiang L, Bullen JW, Zhang C, Samanta D, et al. HIF-1 regulates CD47 expression in breast cancer cells to promote evasion of phagocytosis and maintenance of cancer stem cells. Proc Natl Acad Sci U S A. 2015;112(45):E6215-E6223.
[Google Scholar]
[CrossRef]
|
| 52. |
van Duijn A, Van der Burg SH, Scheeren FA. CD47/SIRPα axis: bridging innate and adaptive immunity. J Immuno Ther Cancer. 2022;10(7):e004589.
[Google Scholar]
[CrossRef]
|
| 53. |
Jacobsson H, Harrison H, Hughes É, Persson E, Rhost S, Fitzpatrick P, et al. Hypoxia-induced secretion stimulates breast cancer stem cell regulatory signalling pathways. Mol Oncol. 2019;13(8):1693-1705.
[Google Scholar]
[CrossRef]
|
| 54. |
Yan Y, He M, Zhao L, Wu H, Zhao Y, Han L, et al. A novel HIF-2α targeted inhibitor suppresses hypoxia-induced breast cancer stemness via SOD2-mtROS-PDI/GPR78-UPR(ER) axis. Cell Death Differ. 2022;29(9):1769-1789.
[Google Scholar]
[CrossRef]
|
| 55. |
Peng F, Wang JH, Fan WJ, Meng YT, Li MM, Li TT, et al. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene. 2018;37(8):1062-1074.
[Google Scholar]
[CrossRef]
|
| 56. |
Fu A, Yao B, Dong T, Chen Y, Yao J, Liu Y, et al. Tumor-resident intracellular microbiota promotes metastatic colonization in breast cancer. Cell. 2022;185(8):1356-1372.e26.
[Google Scholar]
[CrossRef]
|
| 57. |
Ma W, Zhang L, Chen W, Chang Z, Tu J, Qin Y, et al. Microbiota enterotoxigenic Bacteroides fragilis-secreted BFT-1 promotes breast cancer cell stemness and chemoresistance through its functional receptor NOD1. Protein Cell. 2024;15(6):419-440.
[Google Scholar]
[CrossRef]
|
| 58. |
Leontovich AA, Jalalirad M, Salisbury JL, Mills L, Haddox C, Schroeder M, et al. NOTCH3 expression is linked to breast cancer seeding and distant metastasis. Breast Cancer Res. 2018;20(1):105.
[Google Scholar]
[CrossRef]
|
| 59. |
Tian Y, Zhang P, Mou Y, Yang W, Zhang J, Li Q, et al. Silencing Notch4 promotes tumorigenesis and inhibits metastasis of triple-negative breast cancer via Nanog and Cdc42. Cell Death Discov. 2023;9(1):148.
[Google Scholar]
[CrossRef]
|
| 60. |
Zhou L, Wang D, Sheng D, Xu J, Chen W, Qin Y, et al. NOTCH4 maintains quiescent mesenchymal-like breast cancer stem cells via transcriptionally activating SLUG and GAS1 in triple-negative breast cancer. Theranostics. 2020;10(5):2405-2421.
[Google Scholar]
[CrossRef]
|
| 61. |
Kumar S, Nandi A, Singh S, Regulapati R, Li N, Tobias JW, et al. Dll1(+) quiescent tumor stem cells drive chemoresistance in breast cancer through NF-κB survival pathway. Nat Commun. 2021;12(1):432.
[Google Scholar]
[CrossRef]
|
| 62. |
Ma J, Gong Y, Sun X, Liu C, Li X, Sun Y, et al. Tumor suppressor FRMD3 controls mammary epithelial cell fate determination via notch signaling pathway. Sci. Adv. 2024;10(27):eadk8958.
[Google Scholar]
[CrossRef]
|
| 63. |
Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149(6):1192-1205.
[Google Scholar]
[CrossRef]
|
| 64. |
Thacker G, Henry S, Nandi A, Debnath R, Singh S, Nayak A, et al. Immature natural killer cells promote progression of triple-negative breast cancer. Sci Transl Med. 2023;15(686):eabl4414.
[Google Scholar]
[CrossRef]
|
| 65. |
Xiao G, Lu W, Yuan J, Liu Z, Wang P, Fan H. Fbxw7 suppresses carcinogenesis and stemness in triple-negative breast cancer through CHD4 degradation and Wnt/β-catenin pathway inhibition. J Transl Med. 2024;22(1):99.
[Google Scholar]
[CrossRef]
|
| 66. |
Yan L, Wu M, Wang T, Yuan H, Zhang X, Zhang H, et al. Breast Cancer Stem Cells Secrete MIF to Mediate Tumor Metabolic Reprogramming That Drives Immune Evasion. Cancer Res. 2024;84(8):1270-1285.
[Google Scholar]
[CrossRef]
|
| 67. |
Jiang J. Hedgehog signaling mechanism and role in cancer. Seminars in cancer biology. 2022;85:107-122.
[Google Scholar]
[CrossRef]
|
| 68. |
Zhu R, Gires O, Zhu L, Liu J, Li J, Yang H, et al. TSPAN8 promotes cancer cell stemness via activation of sonic Hedgehog signaling. Nat Commun. 2019;10(1):2863.
[Google Scholar]
[CrossRef]
|
| 69. |
Valenti G, Quinn HM, Heynen G, Lan L, Holland JD, Vogel R, et al. Cancer Stem Cells Regulate Cancer-Associated Fibroblasts via Activation of Hedgehog Signaling in Mammary Gland Tumors. Cancer Res. 2017;77(8):2134-2147.
[Google Scholar]
[CrossRef]
|
| 70. |
Cazet AS, Hui MN, Elsworth BL, Wu SZ, Roden D, Chan CL, et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat Commun. 2018;9(1):2897.
[Google Scholar]
[CrossRef]
|
| 71. |
Zhu T, Zheng J, Zhuo W, Pan P, Li M, Zhang W, et al. ETV4 promotes breast cancer cell stemness by activating glycolysis and CXCR4-mediated sonic Hedgehog signaling. Cell Death Discov. 2021;7(1):126.
[Google Scholar]
[CrossRef]
|
| 72. |
Farahzadi R, Valipour B, Fathi E, Pirmoradi S, Molavi O, Montazersaheb S, et al. Oxidative stress regulation and related metabolic pathways in epithelial-mesenchymal transition of breast cancer stem cells. Stem Cell Res Ther. 2023;14(1):342.
[Google Scholar]
[CrossRef]
|
| 73. |
Strietz J, Stepputtis SS, Follo M, Bronsert P, Stickeler E, Maurer J. Human Primary Breast Cancer Stem Cells Are Characterized by Epithelial-Mesenchymal Plasticity. Int J Mol Sci. 2021;22(4):1808.
[Google Scholar]
[CrossRef]
|
| 74. |
Meyer F, Engel AM, Krause AK, Wagner T, Poole L, Dubrovska A, et al. Efficient DNA Repair Mitigates Replication Stress Resulting in Less Immunogenic Cytosolic DNA in Radioresistant Breast Cancer Stem Cells. Front Immunol. 2022;13:765284.
[Google Scholar]
[CrossRef]
|
| 75. |
Sultan M, Vidovic D, Paine AS, Huynh TT, Coyle KM, Thomas ML, et al. Epigenetic Silencing of TAP1 in Aldefluor(+) Breast Cancer Stem Cells Contributes to Their Enhanced Immune Evasion. Stem Cells. 2018;36(5):641-654.
[Google Scholar]
[CrossRef]
|
| 76. |
Folberg R, Maniotis AJ. Vasculogenic mimicry. APMIS. 2004;112(7–8):508-525.
[Google Scholar]
[CrossRef]
|
| 77. |
Hori A, Shimoda M, Naoi Y, Kagara N, Tanei T, Miyake T, et al. Vasculogenic mimicry is associated with trastuzumab resistance of HER2-positive breast cancer. Breast Cancer Res. 2019;21(1):88.
[Google Scholar]
[CrossRef]
|
| 78. |
Xu J, Yang X, Deng Q, Yang C, Wang D, Jiang G, et al. TEM8 marks neovasculogenic tumor-initiating cells in triple-negative breast cancer. Nat Commun. 2021;12(1):4413.
[Google Scholar]
[CrossRef]
|
| 79. |
Liu TJ, Sun BC, Zhao XL, Zhao XM, Sun T, Gu Q, et al. CD133+ cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple-negative breast cancer. Oncogene. 2013;32(5):544-553.
[Google Scholar]
[CrossRef]
|
| 80. |
Liu T, Sun B, Zhao X, Li Y, Zhao X, Liu Y, et al. USP44+ Cancer Stem Cell Subclones Contribute to Breast Cancer Aggressiveness by Promoting Vasculogenic Mimicry. Mol Cancer Ther. 2015;14(9):2121-2131.
[Google Scholar]
[CrossRef]
|
| 81. |
Liu H, Wang H, Chen D, Gu C, Huang J, Mi K. Endoplasmic reticulum stress inhibits 3D Matrigel-induced vasculogenic mimicry of breast cancer cells via TGF-β1/Smad2/3 and β-catenin signaling. FEBS Open Bio. 2021;11(9):2607-2618.
[Google Scholar]
[CrossRef]
|
| 82. |
Das S, Mukherjee P, Chatterjee R, Jamal Z, Chatterji U. Enhancing Chemosensitivity of Breast Cancer Stem Cells by Downregulating SOX2 and ABCG2 Using Wedelolactone-encapsulated Nanoparticles. Mol Cancer Ther. 2019;18(3):680-692.
[Google Scholar]
[CrossRef]
|
| 83. |
Dean M. ABC transporters, drug resistance, and cancer stem cells. J Mammary Gland Biol Neoplasia. 2009;14(1):3-9.
[Google Scholar]
[CrossRef]
|
| 84. |
Yang YS, Jia XZ, Lu QY, Cai SL, Huang XT, Yang SH, et al. Exosomal DEK removes chemoradiotherapy resistance by triggering quiescence exit of breast cancer stem cells. Oncogene. 2022;41(18):2624-2637.
[Google Scholar]
[CrossRef]
|
| 85. |
Wang T, Fahrmann JF, Lee H, Li YJ, Tripathi SC, Yue C, et al. JAK/STAT3-Regulated Fatty Acid β-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab. 2018;27(1):136-150.e5.
[Google Scholar]
[CrossRef]
|
| 86. |
Qin S, He X, Lin H, Schulte BA, Zhao M, Tew KD, et al. Nrf2 inhibition sensitizes breast cancer stem cells to ionizing radiation via suppressing DNA repair. Free Radic Biol Med. 2021;169:238-247.
[Google Scholar]
[CrossRef]
|
| 87. |
Yu L, Zang C, Ye Y, Liu H, Eucker J. Effects of BYL-719 (alpelisib) on human breast cancer stem cells to overcome drug resistance in human breast cancer. Front pharmacol. 2024;15:1443422.
[Google Scholar]
[CrossRef]
|
| 88. |
Mediratta K, El-Sahli S, Marotel M, Awan MZ, Kirkby M, Salkini A, et al. Targeting CD73 with flavonoids inhibits cancer stem cells and increases lymphocyte infiltration in a triple-negative breast cancer mouse model. Front immunol. 2024;15:1366197.
[Google Scholar]
[CrossRef]
|
| 89. |
Ding L, Jiang H, Li Q, Li Q, Zhang TT, Shang L, et al. Ropivacaine as a novel AKT1 specific inhibitor regulates the stemness of breast cancer. J Exp Clin Cancer Res. 2024;43(1):90.
[Google Scholar]
[CrossRef]
|
| 90. |
Hirata N, Yamada S, Yanagida S, Ono A, Kanda Y. FTY720 Inhibits Expansion of Breast Cancer Stem Cells via PP2A Activation. Int J Mol Sci. 2021;22(14):7259.
[Google Scholar]
[CrossRef]
|
| 91. |
Hsieh HY, Chuang HC, Shen FH, Detroja K, Hsin LW, Chen CS. Targeting breast cancer stem cells by novel HDAC3-selective inhibitors. Eur J Med Chem. 2017;140:42-51.
[Google Scholar]
[CrossRef]
|
| 92. |
Alraouji NN, Al-Mohanna FH, Ghebeh H, Arafah M, Almeer R, Al-Tweigeri T, et al. Tocilizumab potentiates cisplatin cytotoxicity and targets cancer stem cells in triple-negative breast cancer. Mol Carcinog. 2020;59(9):1041-1051.
[Google Scholar]
[CrossRef]
|
| 93. |
Ly S, Anand V, El-Dana F, Nguyen K, Cai Y, Cai S, et al. Anti-GD2 antibody dinutuximab inhibits triple-negative breast tumor growth by targeting GD2(+) breast cancer stem-like cells. J Immunother Cancer. 2021;9(3).
[Google Scholar]
[CrossRef]
|
| 94. |
Yan J, Xie Y, Liu Z, Yang Y, Zhou T. DLL4-targeted CAR-T therapy sensitizes neoadjuvant chemotherapy via eliminating cancer stem cells and reshaping immune microenvironment in HER2(+) breast cancer. J Immunother Cancer. 2024;12(11).
[Google Scholar]
[CrossRef]
|
| 95. |
Byrd TT, Fousek K, Pignata A, Szot C, Samaha H, Seaman S, et al. TEM8/ANTXR1-Specific CAR T Cells as a Targeted Therapy for Triple-Negative Breast Cancer. Cancer Res. 2018;78(2):489-500.
[Google Scholar]
[CrossRef]
|
| 96. |
Doheny D, Sirkisoon S, Carpenter RL, Aguayo NR, Regua AT, Anguelov M, et al. Combined inhibition of JAK2-STAT3 and SMO-GLI1/tGLI1 pathways suppresses breast cancer stem cells, tumor growth, and metastasis. Oncogene. 2020;39(42):6589-6605.
[Google Scholar]
[CrossRef]
|
| 97. |
Scatena C, Roncella M, Di Paolo A, Aretini P, Menicagli M, Fanelli G, et al. Doxycycline, an Inhibitor of Mitochondrial Biogenesis, Effectively Reduces Cancer Stem Cells (CSCs) in Early Breast Cancer Patients: A Clinical Pilot Study. Front Oncol. 2018;8:452.
[Google Scholar]
[CrossRef]
|
| 98. |
Zhang S, Zhang H, Ghia EM, Huang J, Wu L, Zhang J, et al. Inhibition of chemotherapy resistant breast cancer stem cells by a ROR1 specific antibody. Proc Natl Acad Sci U S A. 2019;116(4):1370-1377.
[Google Scholar]
[CrossRef]
|
| 99. |
Goldstein LJ, Mansutti M, Levy C, Chang JC, Henry S, Fernandez-Perez I, et al. A randomized, placebo-controlled phase 2 study of paclitaxel in combination with reparixin compared to paclitaxel alone as front-line therapy for metastatic triple-negative breast cancer (fRida). Breast Cancer Res Treat. 2021;190(2):265-275.
[Google Scholar]
[CrossRef]
|

 ,
Suling Liu
1,2,3
,
Suling Liu
1,2,3